Interstitial lung diseases (ILDs) are a heterogeneous group of disorders characterized by inflammation and/or fibrosis.1 Pulmonary fibrosis develops due to repeated cycles of injury and impaired repair with fibroblast activation and migration with the resultant deposition of extracellular matrix components.2,3 This injury may be mediated by environmental, infectious or immune-mediated pathways. Over time, these pathways can become self-sustaining, resulting in progressive fibrosis.4
There are several different types of ILDs based on their aetiology, and understanding the underlying pathogenesis and clinical differences between these conditions is crucial for early treatment. Idiopathic pulmonary fibrosis (IPF) is considered the prototypical progressive fibrosing lung disease.5 However, a proportion of non-IPF ILDs can develop a progressive fibrotic phenotype with a prognosis similar to IPF.6 These include hypersensitivity pneumonitis (HP), connective tissue disease-associated ILD (CTD-ILD), organizing pneumonia (OP), sarcoidosis, idiopathic nonspecific interstitial pneumonia (NSIP) and unclassifiable ILD.7 In this review, we discuss the recent updates in the diagnosis and management of fibrotic interstitial lung diseases (fILDs), including the new definition of progressive pulmonary fibrosis (PPF).8
Diagnosis
The determination of the aetiology of ILDs (Figure 1) is vital to treatment and prognostication. The initial workup includes a detailed history, including exposures, medications and family history; physical examination findings; pulmonary function testing (PFT); serologies and high-resolution computed tomography (HRCT) scan.9 Depending on the results of these tests, other diagnostic modalities, such as bronchoscopy with bronchoalveolar lavage (BAL) and transbronchial forceps biopsies, including genomic classifier (GC), transbronchial lung cryobiopsies (TBLCs) and/or surgical lung biopsies (SLBs), may be necessary.8 Multidisciplinary discussion (MDD) with a team of pulmonologists, thoracic radiologists and thoracic pathologists is a key component in the diagnosis and management of ILD.9–11
Figure 1: Aetiologies of interstitial lung diseases
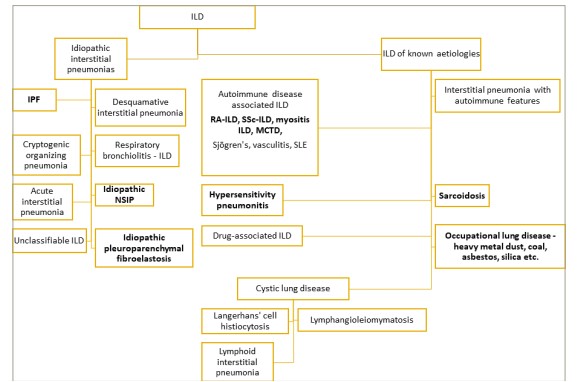
Diseases prone to progressive fibrosis are indicated in bold.
ILD = interstitial lung disease; IPF = idiopathic pulmonary fibrosis; MCTD = mixed connective tissue disease; NSIP = nonspecific interstitial pneumonia; RA-ILD = rheumatoid arthritis-related interstitial lung disease; SLE = systemic lupus erythematosus; SSc-ILD = scleroderma interstitial lung disease.
High-resolution computed tomography scan
Fibrotic ILD is characterized by evidence of fibrosis, such as honeycombing or traction bronchiectasis on imaging involving at least 10% of the lungs or on pathology.12,13 Usual interstitial pneumonia (UIP) is described as having basal predominance, peripheral reticulations with honeycombing and traction bronchiectasis. In contrast, NSIP is a homogeneous disease with diffuse ground-glass opacities and subpleural sparing. The pattern of OP is polymorphous, but commonly shows patchy opacities that may be peripheral or peribronchovascular.14,15 Figures 2 and 3 summarize common computed tomography patterns associated with the various ILDs.
Figure 2: Key pattern characteristics on high-resolution computed tomography scans
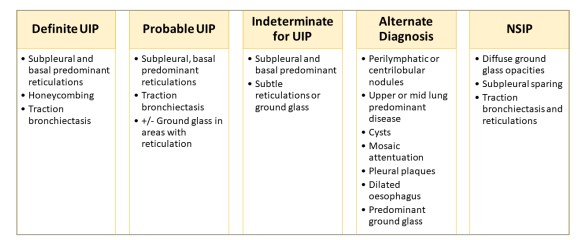
NSIP = nonspecific interstitial pneumonia; UIP = usual interstitial pneumonia.
Figure 3: Common imaging patterns associated with different interstitial lung diseases
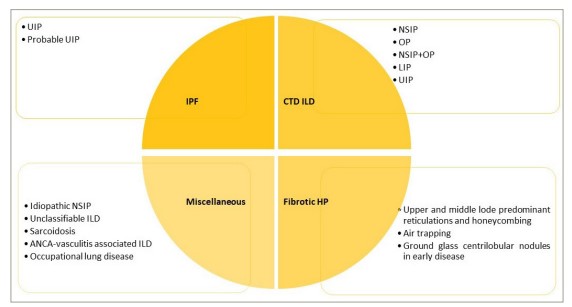
ANCA = anti-neutrophil cytoplasmic antibodies; CTD = connective tissue disease; HP = hypersensitivity pneumonitis; ILD = interstitial lung disease; IPF = idiopathic pulmonary fibrosis; LIP = lymphoid interstitial pneumonia; NSIP = nonspecific interstitial pneumonia; OP = organizing pneumonia; UIP = usual interstitial pneumonia.
Serologies
Although most connective tissue diseases (CTDs) can be associated with pulmonary manifestations, the diagnostic criteria for CTDs, except scleroderma, do not include the presence of ILD.16 Serologic testing can help identify the aetiology of CTD-ILD or the presence of interstitial pneumonia with autoimmune features for those who may not meet all the criteria for CTD but have suggestive features. The current guidelines from the American Thoracic Society (ATS), European Respiratory Society (ERS), Japanese Respiratory Society (JRS), and Latin American Thoracic Society (ALAT) recommend serologic testing to exclude CTD as a possible aetiology. While the practice patterns regarding specific testing varied among panellists, a complete panel as listed in Table 1 is recommended.9
Table 1: Serologic testing in interstitial lung diseases
|
Antibodies |
Extrapulmonary manifestations |
Associated CTD |
|
RF and anti-CCP |
Polyarthritis, joint deformities, arthralgia and rheumatoid nodules |
Rheumatoid arthritis |
|
Anti-Scl70, anti-centromere, anti-RNA polymerase III, CENP-A, CENP-B, RP11, RP155, U1-snRNP RNP A, U1-snRNP RNP C, U1-snRNP RNP-70kd, fibrillarin, Th/To, PM/Scl-100 and PM/Scl-75 |
Raynaud’s phenomenon, skin tightening, trismus, telangiectasias and capillary dropout |
Systemic sclerosis |
|
SSA and SSB |
Xerophthalmia and xerostomia |
Sjögren’s syndrome |
|
Anti-tRNA synthetase Jo-1, PL7, PL12, EJ, OJ, KS Anti-MDA5 (CADM140) Anti-Mi2 SSA-52kD Anti-HMG CoR CK and aldolase |
Proximal muscle weakness, mechanic’s hands and shawl sign |
Polymyositis/dermatomyositis/anti-synthetase syndrome, necrotizing autoimmune myositis |
|
Anti-dsDNA Anti-Smith |
Cytopenia, rash, nephritis, neuropsychiatric symptoms, polyarthralgia, serositis and constitutional symptoms |
Systemic lupus erythematosus |
|
Anti-U1-RNP Anti-Ku Anti-PM/Scl75 ANA ≥1:320 |
Raynaud phenomenon, arthritis, puffy fingers, sclerodactyly, serositis and oesophageal dysmotility |
Mixed connective tissue disease or overlap syndromes |
ANA = anti-nuclear antibodies;anti-PM/Scl = anti-polymyositis/sclerderma ;anti-RP = anti-RNA polymerase antibodies;anti-Th/To = antibodies to Th/To ribonucleoprotein ;CADM = clinically amyopathic dermatomyositis;CCP = cyclic citrullinated peptide;CENP = centromeric proteins ;CK = creatinine kinase;CTD = connective tissue disease;dsDNA = double-stranded deoxyribonucleic acid;HMG CoR = 3-hydroxy-2-methylglutaryl-CoA reductase;kD = kilodalton;MDA = melanoma differentiation-associated protein;Mi2 = nucleosome remodeling deacetylase complex;RF = rheumatoid factor;RNA = ribonucleic acid;snRNP = small nuclear ribonucleoprotein;SSA = Sjӧgren’s syndrome antibody A/B;SSB = Sjӧgren’s syndrome antibody B;U1RNP = uridyl-ribonucleoprotiein.
Bronchoalveolar lavage
Differential cell count on the BAL sample may provide additional information on the diagnosis of ILD.17 In healthy individuals, BAL fluid is primarily composed of macrophages (80–90%) and about 5–15% lymphocytes.18 Although studies have shown a difference in cell counts between IPF and non-IPF ILDs, the data are insufficient to utilize within the IPF diagnostic algorithm.9 In patients with clinical presentation suggestive of IPF and with UIP patterns on the HRCT scan, BAL is not recommended.9 BAL is recommended for patients with probable UIP or an indeterminate pattern chest imaging and when alternate diagnosis, particularly HP, is suspected.9,17 Lymphocyte-predominant fluid may be seen most commonly in cellular HP but can also be present with CTD-ILD or sarcoidosis.19 The results may be equivocal in fibrotic HP, and a detailed exposure history is vital to the diagnosis. The overall risks of bronchoscopy are minimal but should be carefully considered in individuals with significant resting hypoxia.20
Histopathology
While history, physical examination, imaging and serology can provide a diagnosis in most cases of ILD, sometimes a definitive diagnosis is not apparent. Biopsies may be required to further inform decision making. Over the years, several techniques have evolved, each with its own limitations and advantages.
Transbronchial lung biopsy
Transbronchial lung biopsies (TLBs) are performed using flexible forceps, are minimally invasive procedures and do not require general anaesthesia. This technique is a safe and accessible modality.21,22 The diagnostic yield can vary from 30% to 75%, and TLBs show poor concordance with transbronchial cryobiopsy and SLB.22,23 In general, it does not have utility in the diagnosis of most ILDs. The genomic classifier (GC) combines RNA sequencing and machine learning algorithms to better phenotype tissues obtained on transbronchial biopsies.24 GC has a specificity of 92% and a sensitivity of 68% in predicting UIP patterns of fibrosis. While guidelines do not make any recommendations for or against the use of GC, this may aid diagnosis in select patients.8,24
Surgical lung biopsy
Video-assisted thoracoscopic surgery is the preferred approach in patients well enough to tolerate single-lung ventilation. The diagnostic yield of SLB is estimated at 90%, and the risk of pneumothorax and severe bleeding is 6%.9 Biopsies should be obtained from multiple lobes to account for histopathological variation across the lung. SLBs carry the risk of acute exacerbations and even death; every patient should undergo MDD and be referred for SLB only when noninvasive measures have been inconclusive.9,25 Severe physiological impairment can limit the safety of SLB; the current ATS/ERS/JRS/ALAT guidelines suggest TBLC as an alternative in these cases.8
Transbronchial lung cryobiopsy
The diagnostic yield of TBLC across studies has been about 79% in patients with undetermined ILD and has around 70% diagnostic agreement with SLB.26–28 Current guidelines recommend considering TBLC at an experienced center when available, with emphasis on the need for pulmonologist and thoracic pathologist’s expertise. Percent predicted forced vital capacity (ppFVC) <50%, per cent predicted diffusion capacity for carbon monoxide (ppDLCO) <35%, moderate-to-severe pulmonary hypertension (estimated pulmonary systolic pressure >40 mmHg by echocardiography suggestive of although not diagnostic for pulmonary hypertension), other echocardiographic signs of pulmonary hypertension (tricuspid regurgitant velocity >2.8 m/s), significant hypoxaemia and uncorrectable bleeding are risk factors for complications related to TBLC.8,29–31
Disease monitoring and progression
Most clinical trials have used PFT and HRCT scans to monitor fILDs.13,32–36 The frequency of imaging and PFTs requires an individualized approach.37 Currently, PFTs are recommended at a 3–6-month interval for all fILDs; forced vital capacity (FVC) and diffusion capacity for carbon monoxide (DLCO) are important indicators of disease progression.8,10,38 Serial testing of functional status including a 6-monthly 6-minute walk test would determine disease progression.37 Many biomarkers are involved in various mechanistic pathways of fibrotic diseases, including immunity, epithelial dysfunction and aberrant remodeling. These have shown a correlation with disease outcomes but are not yet validated for use in clinical practice.39,40
The decline in FVC in patients with IPF has been associated with increased mortality and worsening quality of life. Progressive fibrosis is strongly linked to increased morbidity and mortality in patients with ILD.41–46 Around 18–36% of non-IPF ILDs develop a progressive phenotype.47–49 The timely and accurate diagnosis of PPF is important for the initiation of appropriate treatment strategies and prognostication. The comparison of IPF and non-IPF ILD with the progressive fibrotic phenotype showed a similar decline in FVC and disease progression, which was associated with increased mortality.50 Disease progression in PPF attributable to fibrosis is defined as having two of the following three criteria:8
-
Worsening respiratory symptoms
-
Physiological evidence of disease progression
-
Absolute decline in ppFVC by ≥ 5% over 1 year of follow-up
-
Absolute decline in ppDLCO of ≥ 10% within 1 year of follow-up
-
-
Radiological signs of progression
-
Increased traction bronchiectasis
-
New ground glass with traction bronchiectasis
-
Increased reticular abnormalities
-
New or increased honeycombing
-
Volume loss.
-
A decline in FVC should prompt an investigation to determine if fibrosis is truly the cause of lung function decline or if other factors, such as infections, pulmonary hypertension or other cardiac diseases, are contributing to this decline. There are several risk factors associated with the progression of fibrotic ILD as shown in Table 2.12
Table 2: Risk factors for progressive fibrosing ILD
|
Patient factors |
Disease-related factors |
|
Low BMI |
Low baseline FVC and DLCO |
|
Smoking history |
Extent of fibrosis on HRCT |
|
Older age |
Among CTDs, RA-ILD and SSc-ILD |
|
|
Fibrotic HP, unknown antigen |
|
|
Usual interstitial pneumonia pattern on HRCT |
|
|
Desaturation during six-minute walk test |
BMI = body mass index;DLCO = diffusion capacity for carbon monoxide;FVC = forced vital capacity;HP = hypersensitivity pneumonitis;HRCT = high-resolution computed tomography;RA-ILD = rheumatoid arthritis-related interstitial lung disease;SSc-ILD = scleroderma interstitial lung disease.
Pharmacological management
Idiopathic pulmonary fibrosis
The current recommendation is to manage IPFs with a combination of pharmacological and non-pharmacological interventions.8–10 Pharmacotherapy in IPF primarily involves the initiation of antifibrotic therapies (AFTs), which have been found to be safe with long-term use.51,52 Two AFT drugs, pirfenidone and nintedanib, have been shown to decrease the rate of FVC decline in patients with IPF in randomized controlled trials (RCTs).53–56 In the ASCEND trial (A randomized, double-blind, placebo controlled, phase 3 study of the efficacy and safety of pirfenidone in patients with idiopathic pulmonary fibrosis; ClinicalTrials.gov identifier: NCT01366209), pirfenidone demonstrated a 47.9% relative reduction in patients with a 10% FVC decline or death at 52 weeks compared with controls.54 INPULSIS 1 and 2 (A 52 weeks, double blind, randomized, placebo-controlled trial evaluating the effect of oral BIBF 1120, 150 mg twice daily, on annual forced vital capacity decline, in patients with idiopathic pulmonary fibrosis [IPF]; Clinical Trials.gov identifier: NCT01335464,) showed a between-group difference of 125.3 mL and 93.7 mL annual FVC change, respectively.53 While both have shown some benefit in limiting acute exacerbations of IPF, this has been less consistent across studies.53–56 Pooled data from the various RCTs for these therapies suggest improved survival and reduced respiratory-related hospitalizations with AFTs, thus improving the life expectancy of patients with this devastating disease.57–60
While these AFTs have certainly changed the landscape of the treatment of IPF, there are concerns about poor tolerability in the elderly IPF patient population. Pirfenidone can be commonly associated with gastrointestinal (GI) events, including nausea, vomiting, diarrhoea, gastro-oesophageal reflux disease (GORD) and skin-related adverse effects such as rash and photosensitivity.54,55 Nintedanib can be associated with GI side effects, including diarrhoea, nausea and vomiting. Poor appetite and weight loss were reported with both these drugs.53,56 Both AFTs can cause elevation in transaminase levels, and serial monitoring is recommended for patients on therapy. Given the similar efficacy of both AFTs and the lack of biomarkers to suggest a better response in an individual to either drug, discussion of the potential benefits and side effects and involving the patient and caregiver in the decision on which AFT to initiate are crucial. Further discussions regarding the tolerability and, if necessary, dose adjustments should made at every visit.61,62
Non-idiopathic pulmonary fibrosis fibrotic interstitial lung diseases (non-IPF fILDs)
Immunomodulatory therapies
In CTD-ILDs, immunosuppression is the cornerstone of therapy, with varying levels of evidence for different therapeutic agents. Commonly used agents include cyclophosphamide (CYC), mycophenolate mofetil (MMF), rituximab (RTX), azathioprine (AZA), tocilizumab, methotrexate and prednisone.63 Scleroderma-ILD has been the most investigated CTD; approach to immunosuppression for other CTD-ILDs is largely extrapolated from these data. Scleroderma Lung Study I (Cyclophosphamide versus placebo in scleroderma lung study; ClinicalTrials.gov identifier: NCT00004563) and II (A randomized controlled trial to compare the efficacy of oral mycophenolate mofetil with placebo in patients with systemic sclerosis related early interstitial lung disease; ClinicalTrials.gov identifier: NCT02896205) reported improvement in ppFVC with both MMF and CYC; MMF has become the preferred agent due to a favourable adverse effect profile and similar efficacy compared with CYC.64,65 Monoclonal antibodies including RTX and tocilizumab have been investigated for the management of systemic sclerosis (SSc).66–69 While tocilizumab did not meet its primary endpoint of improving the modified Rodnan skin score (mRSS), ppFVC was stabilized in patients with early SSc-ILD and elevated acute-phase reactants.69 RTX has shown similar efficacy to CYC, in terms of time to treatment failure and progression-free survival, in patients with severe or progressive CTD-ILDs, with fewer adverse effects.67 RTX has also been effective in improving mRSS and ppFVC compared with placebo in patients with SSc.68 Guidelines have not standardized the treatment algorithm; MMF is preferred as the initial agent for CTD-ILDs with or without corticosteroids, while RTX and CYC are considered rescue therapies. Monitoring for hepatic, renal and bone marrow functions and opportunistic infections is required while on immunomodulatory therapies.
Corticosteroids have shown improvement in FVC and DLCO in patients with nonfibrotic HP,
but not with predominantly fibrotic HP (fHP).70 One study has shown that treatment of fHP with MMF or AZA is associated with improvement in DLCO but not in FVC.71 However, a shorter telomere length was associated with lower FVC in fHP patients exposed to MMF, similar to IPF patients, and should be considered cautiously.38
Antifibrotics
More recently, AFTs have been investigated in patients with non-IPF fILDs as an additional therapy to immunosuppression and with PPF. The SENSCIS trial (A double blind, randomised, placebo-controlled trial evaluating efficacy and safety of oral nintedanib treatment for at least 52 weeks in patients with systemic sclerosis associated interstitial lung disease [SSc-ILD]; ClinicalTrials.gov identifier: NCT02597933) showed that nintedanib was associated with a significantly lower annual FVC decline compared with placebo in patients with SSc-ILD on a stable background therapy, and should be considered, especially for patients with predominantly fibrotic presentation.13 Side effects were similar to those in the IPF trials, irrespective of background therapy. The LOTUSS trial (The LOTUSS trial: An open-label, randomized, phase 2 study of the safety and tolerability of pirfenidone when administered to patients with systemic sclerosis-related interstitial lung disease [SSc-ILD] [LOTUSS]; ClinicalTrials.gov identifier: NCT01933334) reported that the use of pirfenidone in addition to background immunosuppression was safe and effective in SSc-ILD, but further investigation is recommended.72 Primarily, GI symptoms including nausea, GORD, fatigue and headache were reported, and tolerability was not affected by concomitant MMF.72
The INBUILD trial (A double blind, randomized, placebo-controlled trial evaluating the efficacy and safety of nintedanib over 52 weeks in patients with progressive fibrosing interstitial lung disease [PF-ILD]; ClinicalTrials.gov identifier: NCT02999178) showed 57% less FVC decline in patients with progressive ILD with the addition of nintedanib when compared with placebo, with the caveat being that study participants were not on background immunosuppression in the initial 6 months.33 However, the data from the SENSCIS trial can be taken into consideration when deciding therapeutic strategies in patients with CTD-ILD on immunosuppression13 . The fILDs included in this study were fHP, CTD-ILD and unclassifiable ILD among others. Pirfenidone has shown efficacy in reducing FVC decline in non-IPF progressive fILDs, and while it did not meet its primary endpoint of home spirometry stabilization in unclassifiable ILD, the in-office spirometry results were in favour of pirfenidone.35,36 This may be considered an alternate antifibrotic among patients intolerant to nintedanib; guidelines do recommend that further studies evaluating the efficacy of pirfenidone in non-IPF ILD are necessary.8 Although the inclusion criteria for these studies for PPF varied from the current guidelines, criteria recommended per the current guidelines are applicable to clinical practice.8,33–35 Most trials on antifibrotics have used FVC as their primary endpoints, and meta-analysis shows a reduction in all-cause mortality and acute exacerbations in IPF.73,74
Among patients with non-IPF fILDs, particularly, CTD-related ILD and PPF, the question of whether to increase immunosuppression or initiate AFT or both is often raised. While there are no validated biomarkers to inform treatment decisions, several factors including underlying aetiology, extrapulmonary symptoms, HRCT findings, comorbidities and drug tolerability should be taken into consideration. A patient-centred approach with shared decision making and an MDD are optimal for this decision process. Sequential addition of therapy rather than concomitant therapy is advised to enable understanding both efficacy and tolerability of treatment escalation.12 Figure 4 depicts the approach to disease progression in non-IPF ILDs and recommended treatment strategies.
Figure 4: Workup of progressive fibrotic interstitial lung diseases
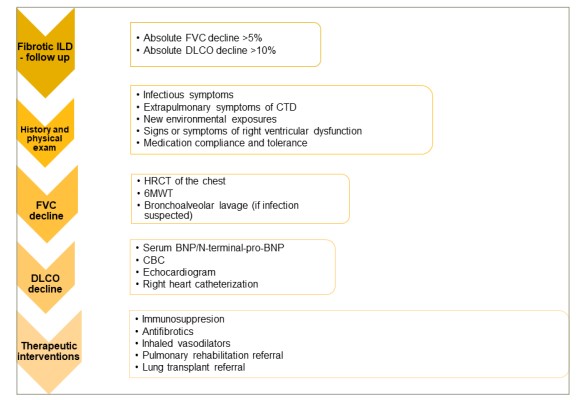
6MWT = 6-minute walk test; BNP = brain natriuretic peptide; CBC = complete blood count; CTD = connective tissue disease; DLCO = diffusion capacity for carbon monoxide; FVC = forced vital capacity; HRCT = high-resolution computed tomography; ILD = interstitial lung disease; NT-pro-BNP = N-terminal pro-brain natriuretic peptide.
Management of comorbidities in fibrotic interstitial lung diseases
Pulmonary hypertension
Pulmonary hypertension due to interstitial lung disease (PH-ILD) has been associated with increased mortality, reduced exercise tolerance and higher supplemental oxygen needs in patients with interstitial lung diseases. Its prevalence ranges from 3% to 86%, with an increasing risk of PH-ILD in more advanced diseases.75–77 PH-ILD should be suspected in patients with hypoxia or DLCO reduction disproportionate to underlying lung diseases. Most cases of PH-ILD are non-severe, with pulmonary vascular resistance ≤5 Woods units.78 The presence of PH-ILD is of great prognostic significance and is an indication for transplant referral if a patient is eligible.79 Studies of vasodilator therapies had been largely disappointing until a recent trial of inhaled treprostinil showed promise as a therapeutic agent, with improved 6-min walk distances and a lower risk of clinical worsening.80 The most commonly reported side effects were cough, headache, dyspnoea, dizziness, nausea, fatigue and diarrhoea. The current European Society of Cardiology (ESC)/European Respiratory Society (ERS) guidelines on the management of PH-ILD recommend supplemental oxygen and noninvasive ventilation when appropriate and state that inhaled treprostinil may be considered for patients with PH-ILD.81
Gastro-oesophageal reflux disease
The recent update of the ATS/ERS/JRS/ALAT guidelines made a conditional recommendation to not treat asymptomatic GORD for the purposes of improving respiratory outcomes due to a lack of evidence supporting improved mortality or lung function decline.8,82,83 The guidelines also suggested avoiding referral for anti-reflux surgery for respiratory outcomes due to a similar lack of evidence.8,84,85 However, almost 90% of patients with IPF can experience GORD, and the incidence can vary in non-IPF ILDs due to disease-specific aetiologies, and treatment with anti-reflux medications is recommended for these patients.86,87
Non-pharmacological interventions
Symptom burden, physical activity, hospitalizations and psychological well-being are important patient-related outcomes in the management of ILDs. Patients with ILDs have greater exercise-induced desaturation and respiratory failure compared with patients with chronic obstructive pulmonary disease (COPD).88 Exercise training is a key component of pulmonary rehabilitation. Exercise training should be individualized and can improve exercise tolerance, without actual changes in lung function.89 Pulmonary rehabilitation improves healthcare-related quality of life, which persists for months after therapy.90 Long-term oxygen therapy should be administered in patients with ILD with resting hypoxaemia.10 Patients with ILD should be referred for lung transplantation and palliative care when appropriate.12,91
Lung transplantation
Based on the International Society for Heart and Lung Transplant consensus statement, lung transplantation should be considered if the risk of mortality from lung disease in the next 2 years is >50% and the 5-year post-transplant survival is >80%.91 The recommended approach to lung transplantation for ILD is summarized in Table 3. 91 The real-world feasibility of referral and listing is dependent on resources and the level of expertise. Overall, the 5-year post-transplant survival of patients with CTD-ILD is similar to that of those with IPF; however, a thorough evaluation of extrapulmonary manifestations is important in this population.92,93
Table 3: Recommendations for lung transplant referral and listing
|
Timing for referral |
Timing of listing |
|
|
|
|
|
|
|
|
|
|
DLCO = diffusion capacity for carbon monoxide;FVC = forced vital capacity;UIP = usual interstitial pneumonia.
Conclusion
Fibrotic ILDs, both IPF and non-IPF, are progressive diseases associated with increased morbidity and mortality. Early recognition of clinical and radiographic progression is pivotal to the timely management of these patients. Antifibrotics are the mainstay of treating progressive fibrotic diseases. Comorbidities, such as pulmonary hypertension, ongoing inflammation due to CTD, infection or fluid overload, are important differentials that should be evaluated. The role of genetic testing and biomarkers in identifying at-risk individuals in clinical settings is yet to be determined. Further investigation into the management of PPF is needed.