|
Highlights
|
Diffuse alveolar hemorrhage (DAH), though rare, can be fatal and requires a high index of suspicion as frank hemoptysis may not be a presenting symptom. For a patient in which bilateral pulmonary infiltrates are present, DAH should be in the differential as additional diagnostic evaluations and different treatment is mandated. We present a case of delayed recognition of DAH, initially thought to be a multilobar pneumonia.
Case
A 67-year-old woman, without any significant past medical history, presented to the emergency department with a 1-day history of low-grade fever, cough, and malaise. She had recently been in contact with a relative who was hospitalized with pneumonia. On examination, her temperature was 100.6°F, heart rate was 98, blood pressure 134/86, respiratory rate 22, and oxygen saturation on room air was 89%. Her only notable exam finding was faint bilateral crackles in the posterior lung fields. A chest x-ray revealed hazy perihilar consolidation in both the right upper lobe and right base, worrisome for pneumonia (Figure 1A and B). Her initial white blood cell count was 9.7 K/µL without bands or neutrophil predominance. Her hemoglobin was 13.6 g/dL, and creatinine was 0.53 mg/dL.
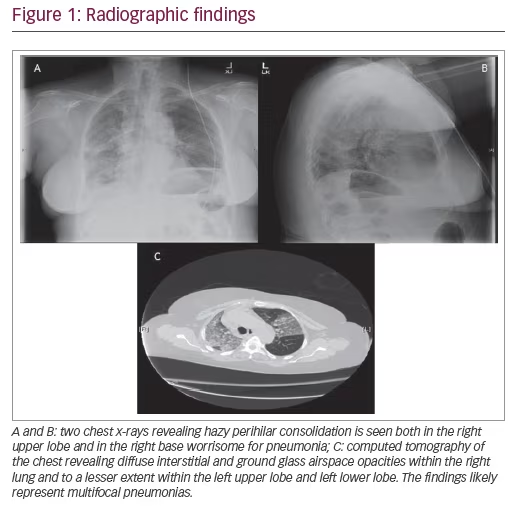
She was admitted to the general medicine ward and started on antibiotics (ceftriaxone and azithromycin) for community acquired pneumonia. The following day, her oxygen requirement continued to increase and she required a 60% large-reservoir oxygen mask to keep her oxygen saturation >92%. A chest computed tomography (CT) scan was ordered, which revealed diffuse interstitial and ground glass airspace opacities within the right lung, lesser extent within the left upper lobe and left lower lobe. The findings likely represent multifocal pneumonias (Figure 1C). Due to continued deterioration the patient’s case was transferred from the hospitalist to the intensivist in the intensive care unit (ICU) when she required a 100% non-rebreather mask. She was intubated immediately and placed on mechanical ventilation. Due to her rapid decline in respiratory status and failure to respond to appropriate antibiotics, a bronchoscopy with bronchoalveolar lavage (BAL) was performed of the bilateral lower lobes with serial aliquots to evaluate for hemorrhage, eosinophils, the airways, and infectious pathogens. In the right lower lobe her red blood cell counts were 1) 50 K/µL; 2) 70 K/µL; and 3) 79 K/µL. In the left lower lobe her red blood cell counts were 1) 19 K/µL; 2) 30 K/µL; and 3) 80 K/µL. She was started on methylprednisolone 500 mg intravenously every 12 hours given that the BAL findings were consistent with DAH. Within 3 days, the patient improved to the point that she was extubated and placed on nasal cannula, and was subsequently discharged from the ICU.
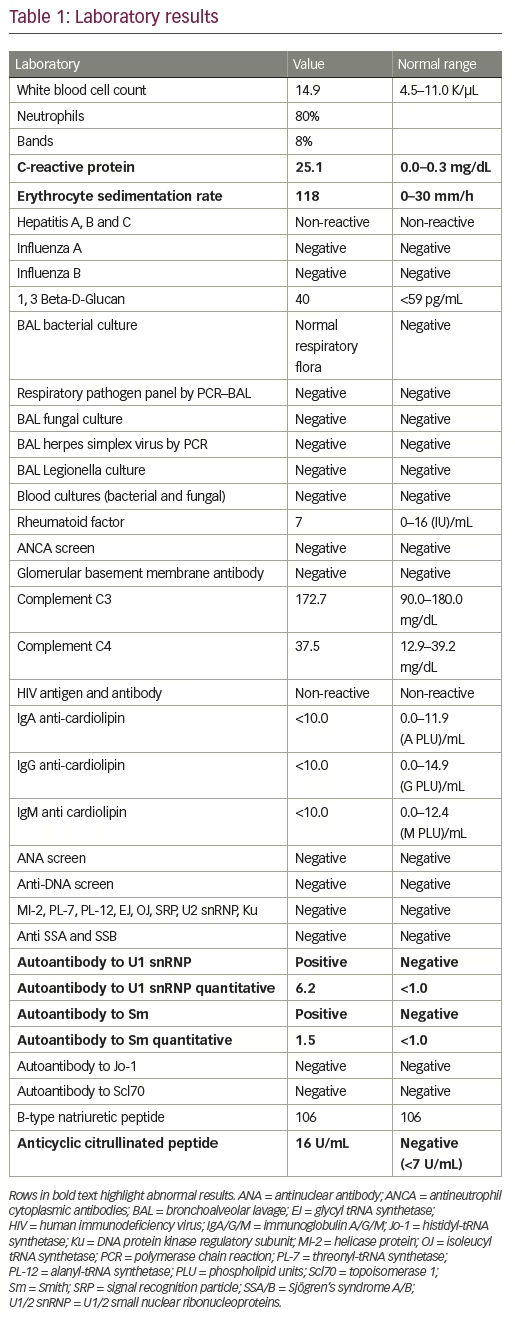
Her evaluation was extensive (Table 1) and only significant for a positive cyclic citrullinated peptide antibody 16 U/mL (negative <7), autoantibody to U1 snRNP 6.2 (negative <1.0), and autoantibody to Smith 1.5 (negative <1.5). All blood cultures were negative at 5 days and all respiratory cultures were negative at 8 weeks. The patient was discharged after 13 days in the hospital and instructed to follow up for further evaluation with rheumatology, given findings concerning for an underlying rheumatologic etiology of her DAH. Additionally, she was instructed to continue immunosuppressive therapy.
Discussion
DAH is a rare but serious condition that can quickly lead to death. Diagnosis relies on a strong clinical suspicion, laboratory studies, imaging and pathology. Over 100 different causes of DAH have been identified; however, the etiologies can be divided into three broad categories.1 For the sake of simplicity we will use a slight variation of the categorization of DAH etiologies as described by Thomas et al. and Albelda et al.: renal disease associated, immune complex associated, and disease associated with neither.2,3 The most common renal causes of DAH include those in which anti-glomerular basement membrane antibodies are present such as Goodpasture syndrome, coagulopathy from end-stage renal disease uremia, and idiopathic concentric glomerulonephritis. Common immune complex associated causes of DAH include systemic lupus erythematous, granulomatosis with polyangiitis, cryoglobulinemia, Henoch-Schonlein purpura, mixed connective-tissue disease, and other vasculitic processes. Common causes of DAH not associated with renal disease or immune complex disease include acute respiratory distress syndrome, idiopathic pulmonary hemosiderosis, bleeding disorders, toxins, medications, trauma, congestive heart failure, malignancy, and pulmonary venous hypertension.
Overall incidence of DAH is difficult to estimate as it is usually a complication of another disease entity. One series of 34 autopsies described the most commonly associated conditions as granulomatosis with polyangiitis, Goodpasture syndrome, idiopathic pulmonary hemosiderosis, collagen vascular disease, and microscopic polyangiitis.4
Mortality can be quite high in DAH with reports ranging between 20–50% while in the hospital.1 Mortality was found to be 60% in patients with DAH from granulomatosis with polyangiitis, the most common cause of DAH.5 One study evaluating microscopic polyangiitis found that up to 30% of patients die during the acute hemorrhage, and in those who survived, the 1-year and 5-year survival was 82% and 68%, respectively.6 Additionally, patients who develop DAH rarely have it in isolation and it is usually the result of an ongoing systemic disease. However, there have been case reports of isolated pulmonary capillaritis and DAH in rheumatoid arthritis and mixed connective tissue disease as seen with our patient.7 Diagnosis of rheumatologic conditions presents a unique set of challenges as disease processes frequently overlap and clinicians rely heavily on antibody panels. Ultimately, survival of DAH is based on the severity of the pulmonary hemorrhage and response to treatment of the underlying cause.
Due to the high mortality, a high degree of suspicion is required and early interventions are necessary. However, treatments should be directed at the suspected or known etiology if possible. If infection is suspected antibiotics should be initiated early. For DAH thought to be secondary to an autoimmune etiology, immunosuppression and corticosteroids are first-line treatments. Plasmapheresis should be used in patients with immunoglobulin or immune complex disease—specifically Goodpasture syndrome.
DAH should be considered in patients with diffuse radiographic opacities, abnormal gas exchange, and hemoptysis. It is important to note hemoptysis may be absent in up to 33% of patients with DAH.8 Regardless of presence of hemoptysis, suspicion for DAH should be high in patients with progressive respiratory symptoms and ground glass or consolidative opacities on imaging, especially when associated with a drop in the patient’s hemoglobin. While DAH was not initially considered in our patient, her failure to respond to traditional treatment led to re-evaluation of the etiologies of her illness upon transfer to the ICU, and prompted bronchoscopic evaluation. This case emphasizes the importance of keeping differential diagnoses broad, especially when patients fail to respond to treatment. In our case, the patient was diagnosed with multilobar pneumonia, which in most instances, would have been appropriate; however, failure to challenge the etiology of her disease could have led to tragic consequences.
While there are characteristic findings with pulmonary function testing (specifically increased diffusing capacity for carbon monoxide [DLCO]), most patients are too unstable to complete pulmonary function testing.9 However, DLCO monitoring can be useful in monitoring recurrence in patients with an established diagnosis of DAH, as in patients with anti-glomerular basement membrane disease.9 Diagnosis of DAH hinges on the use of flexible bronchoscopy. Flexible bronchoscopy with serial BAL is the preferred method to identify DAH and exclude infection, eosinophilia, and malignancy. Guided by imaging abnormalities, the bronchoscope is wedged in a subsegmental bronchus and serial BAL is performed using aliquots of 50–60 mL of sterile saline from the same location. DAH is confirmed when analysis of lavage fluid demonstrates increasing red blood cell count on each subsequent aliquot. Additionally, DAH can be diagnosed through the use of Prussian blue staining to identify hemosiderin-laden macrophages.10 Identification of greater than 20% of 200 total macrophages, which stain positive for hemosiderin, is consistent with the diagnosis of DAH.10
Once diagnosis of DAH has been made, or even if highly suspected, laboratory evaluation should be initiated which may help identify the specific etiology. As already mentioned, a key step is excluding infection with microbiologic studies. Basic laboratory evaluation with urinalysis, creatinine, and blood urea nitrogen should be obtained in all patients to evaluate for pulmonary-renal syndrome. Specific antibody testing, staining, and urine drug screen may identify specific underlying causes of DAH. Antineutrophil cytoplasmic antibody (ANCA) positivity can be associated with DAH due to granulomatosis with polyangiitis or microscopic polyangiitis.11 A positive perinuclear ANCA with antimyeloperoxidase specificity on ELISA is consistent with the diagnosis of microscopic polyangiitis or eosinophilic granulomatosis with polyangiitis. On the other hand, cytoplasmic ANCA and anti-proteinase 3 antibodies on ELISA are most consistent with granulomatosis with polyangiitis. Anti-glomerular basement membrane (GBM) antibodies may be identified using ELISA testing and, if positive, suggest anti-GBM antibody disease (Goodpasture’s). Findings consistent with systemic lupus erythematosus including abnormal complement levels, antinuclear antibodies, and anti-double stranded DNA antibodies, may be further evaluated with antihistone antibodies.12 Antiphospholipid syndrome can be identified by positive anticardiolipin antibodies, lupus anticoagulant, and anti-beta-2-glycoprotein I. The presence of antistreptolysin O, hyaluronidase, or anti-DNase B can be suggestive of poststreptococcal glomerulonephritis. A urine drug screen should be performed, as cocaine can be associated with DAH from direct toxicity or levamisole, an agent frequently used to cut cocaine, can cause capillaritis.6
The treatment of DAH, due to its rarity and heterogeneity, has not been extensively studied in clinical trials, except in severe presentations of systemic lupus erythematosus, granulomatosis with polyangiitis, and mixed cryoglobulinemia, from which management has been extrapolated. Initial stages of treatment are focused on supportive care while working on identifying an underlying diagnosis, ruling out infection, discontinuing potential offending medications, and reversal of anticoagulation. Patients with DAH frequently have hypoxemia requiring supplemental oxygen; often hypoxia is severe to the point that noninvasive or invasive mechanical ventilation is necessary. Use of extracorporeal membrane oxygenation has been used in patients with difficult to treat hypoxemic respiratory failure.13
DAH with capillaritis (as seen in systemic vasculitis, anti-glomerular basement membrane [anti-GBM] syndrome, or rheumatic disease) can be life threatening. Early, systemic glucocorticoid therapy is the mainstay of treatment for capillaritis, regardless of cause, as glucocorticoids are a part of regimens for all these diseases. Dosing is based on expert opinion and is typically administered as pulse dosing of methylprednisolone (500–2,000 mg total daily dose) for up to 5 days. Further immunosuppressive therapy with additional agents is based on identification of specific underlying disease, degree of severity of the illness, and expected responsiveness to glucocorticoids. For example, in patients with DAH secondary to granulomatosis with polyangiitis or microscopic polyangiitis concurrent, use of cyclophosphamide and/or rituximab, in addition to pulse dose steroids, is considered common practice.14,15 Furthermore, use of plasma exchange therapy can be considered in patients with ventilator-dependent DAH, although its use in these patients is not based on randomized controlled trials.16 In contrast, in patients with anti-GBM antibody disease, therapy typically consists of plasma exchange, high-dose glucocorticoids, and an immunosuppressive agent.17
Conclusion
Multilobar pneumonia and DAH may present with similar clinical and radiographic characteristics. In the appropriate setting, pneumonias may be associated with increased alveolar bleeding if the pneumonic burden is high. However, it is important to recognize when pulmonary infiltrates, with or without infection, have a presentation of DAH, as the treatment is to provide what may essentially be lifesaving immunosuppression. Additionally, the etiology must be evaluated and addressed as DAH may be a problem, but not the diagnosis. Again, a high index of suspicion is required and immediate immunosuppression may be of utmost importance.