Pulmonary alveolar proteinosis (PAP) is a rare pulmonary syndrome with a prevalence of approximately seven cases per 1 million individuals.1,2 PAP affects people of all age, sex, race and socioeconomic status. Based on the largest population studies conducted in the United States and Japan, the median age for diagnosis in Japan is 51 years with a male:female ratio of 2.1:1.0.2 In a larger population study conducted in the United States, the male:female ratio was reported to be 1:1.17.3 PAP has also been associated with inhalational exposures to environmental toxins, such as cigarette smoke, organic and inorganic dust, and silica.4 The majority of patients with PAP report a history of smoking, and 26% of patients have a history of occupational exposure to dust or particulates.2 In a healthy lung, surfactant regulates the air-liquid interface by lowering surface tension, thus preventing alveolar collapse at end-expiration. Surfactant also helps with killing of pathogens or preventing their dissemination and modulates immune responses.5 Here, we will review the pathophysiology of this rare pulmonary syndrome, its clinical presentations, diagnosis procedures, and current treatment for patients with PAP.
Mechanisms and pathophysiology
Surfactant homeostasis
Surfactant in the lungs primarily reduces surface tension, allowing for efficient gas exchange, and also provides first-line defence against microbial pathogens.6 It is composed of 80% polar phospholipids (predominantly phosphatidylcholine), 10% cholesterol, and 10% surfactant proteins.7,8 Surfactant homeostasis is regulated by type II alveolar epithelial cells, which secrete, recycle and restore surfactant into the alveolar space; and alveolar macrophages, which clear surfactant via catabolism.3
Granulocyte–macrophage colony-stimulating factor (GM-CSF), a glycoprotein cytokine involved in the differentiation of neutrophils and macrophages, has been found to play a crucial role in maintaining surfactant homeostasis. The importance of pulmonary GM-CSF was incidentally discovered in GM-CSF-deficient (Csf2-/-) mice. The mice developed a pathophysiology that was similar to PAP disease in humans, and had no major defect in haematopoiesis.9 GM-CSF activates a signalling pathway that is essential for the differentiation of alveolar macrophages. Any disruption in the pathway results in large, immature, foamy macrophages that are filled with intracytoplasmic lipid droplets, due to dysregulation of cholesterol homeostasis.10 These immature alveolar macrophages are no longer able to efflux cholesterol and clear surfactant, thus leading to surfactant accumulation in the alveoli.10 The ratio of cholesterol:phospholipids in pulmonary surfactant is also markedly increased in both mice and humans with PAP.11
Pathogenesis of primary pulmonary alveolar proteinosis
Primary PAP occurs due to the disruption of the GM-CSF signalling pathway, which leads to the accumulation of surfactant within the alveoli.3,9 There are two forms of primary PAP: autoimmune PAP (aPAP; previously known as idiopathic PAP) and hereditary PAP. The former, aPAP, is associated with elevated anti-GM-CSF autoantibody concentrations.12 GM-CSF autoantibodies are composed of polyclonal immunoglobulin G, have a high binding affinity and can neutralize GM-CSF at higher concentrations than those present physiologically, thus blocking GM-CSF signalling.13 There is a ‘critical threshold’ value of GM-CSF autoantibody concentration. When this value is exceeded, it disrupts the GM-CSF-stimulated functions, such as alveolar macrophages and blood leukocytes, increasing the risk of PAP. Notably, levels of GM-CSF autoantibodies at diagnosis do not appear to correlate with disease severity in individuals with aPAP, and low levels of GM-CSF autoantibodies are also present in those without PAP.14 Hereditary PAP is caused by mutations in the genes that encode the alpha and beta subunits of the GM-CSF receptor, termed CSF2RA and CSF2RB, respectively.15 The prevalence of hereditary PAP is extremely low, accounting for <5% of all PAP cases.1 Interestingly, in family members with identical mutations, the disease severity is variable.16,17
Pathogenesis of secondary pulmonary alveolar proteinosis
Although the pathogenic mechanisms are not well understood, secondary PAP is most often due to reduced numbers or function of alveolar macrophages.18 Systemic disorders, including malignant and non-malignant haematological diseases (most commonly, myelodysplastic syndrome), non-haematological malignancies, immune deficiency syndromes, chronic inflammatory syndromes, chronic infections, and methionyl-tRNA synthetase mutations, are most commonly associated with secondary PAP.3,19
Pathogenesis of congenital pulmonary alveolar proteinosis
Congenital PAP is rare, and is associated with disruptions in the production and function of surfactant.3 These disruptions cause respiratory disease and are associated with pulmonary fibrosis.3 Causes of congenital PAP include mutations in genes required for normal surfactant production, such as surfactant-associated protein B (SFTPB), surfactant-associated protein C (SFTPC), and adenosine triphosphate-binding cassette transporter A3 (ABCA3).3,17,20
Clinical presentation and disease course
Clinical presentation
Many patients with PAP typically present with nonspecific respiratory and systemic symptoms.3 Common symptoms include: dyspnoea on exertion, cough with or without sputum, chest tightness, fatigue, weight loss, and low-grade fever. Physical examination is usually unremarkable; however, crackles and cyanosis have been reported in a small number of patients.3 As PAP is rare, many patients are often misdiagnosed with common pulmonary diseases, such as pneumonia or asthma, before being correctly diagnosed. In these cases, an accurate diagnosis is delayed by an average of 18 months from initial symptom presentation.4
Screening and prevention
PAP is not routinely screened for in newborn babies or adults because of its low prevalence. If PAP is suspected in a paediatric patient, he or she should be screened for mutations that cause hereditary or congenital PAP. Additionally, any patient with ground-glass opacities on imaging, and dyspnoea of insidious onset should have a GM-CSF autoantibody test to screen for aPAP. Currently, there are no proven preventive approaches, but cessation of cigarette smoking and limiting inhalation exposure to environmental toxins could theoretically reduce the risks of aPAP and secondary PAP, respectively.21
Disease course
The clinical course of PAP is heterogeneous and depends on the specific type of PAP. The clinical course of aPAP typically follows one of three patterns: progressive deterioration, stable but unremitting disease, or spontaneous resolution. The clinical course of hereditary PAP is similar to that of aPAP. It has been reported to have a 5-year survival of 95%, if treated with whole lung lavage.22 With secondary PAP, the prognosis is worse, with a reported 2-year survival of 40% and a median survival time of <20 months.23 In some cases, the cause of PAP is unknown. Although it has not been studied prospectively, the decreased survival rate in patients with secondary PAP appears to be linked to the underlying disease, such as a lung infection, immune problems, cancers of the blood system, and exposure to high levels of environmental substances, such as silica or aluminum dust.24 These factors can lead to the development of PAP, rather than the manifestations of PAP itself.25,26 The disease course in patients with congenital PAP is highly dependent on their mutations, and the presentation can range from death within the first hour of life, through to insidious onset of pulmonary fibrosis during childhood or adulthood.27
In a meta-analysis of 343 patients with any PAP-causing disease, the survival rate from the time of diagnosis was 79%, 75% and 68% at 2, 5 and 10 years, respectively.21 The vast majority (94%) of the deaths were directly due to PAP; of those, 72% were due to respiratory failure, 18% were due to uncontrolled infection, and 1.5% were due to cardiac arrest during whole-lung lavage (WLL).21
Pulmonary fibrosis develops in up to 20% of patients with PAP.28 The frequency, aetiology, and pathogenesis remain unclear, but it is associated with a worse prognosis.29
Infections
Patients with PAP are susceptible to secondary infections, including both common and opportunistic pathogens. As a result of impaired alveolar macrophage and neutrophil function, patients have an increased susceptibility to secondary infections.30 Nocardia, atypical mycobacteria, Mycobacterium tuberculosis, fungi (Cryptococcus species in particular), and a host of other microorganisms have been reported to cause the majority of pulmonary and extrapulmonary infections.31 Infection accounts for approximately 20% of deaths in PAP,21 and pathogens including Aspergillus,2 other fungal species, Nocardia, and Mycobacterium species found within 16 months of PAP diagnosis, are associated with poor prognosis and increased mortality.31
Testing and diagnosis
Radiological findings
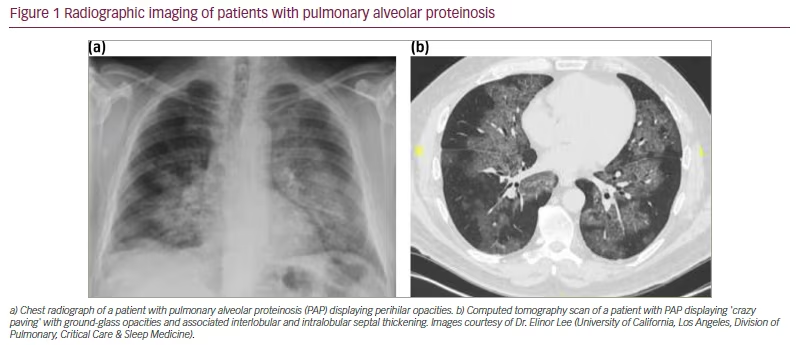
Chest radiographs classically show a ‘bat-wing’ appearance with perihilar lung involvement and sparing of the costophrenic angles (Figure 1aA).12 Computed tomography routinely shows ground-glass opacities with interlobular and intralobular septal thickening in a polygonal pattern similar to cobblestones on pavement, commonly referred to as ‘crazy paving’ (Figure 1bB). This finding, however, is non-specific and found in other pulmonary conditions including infectious, neoplastic and inhalational disorders of the lung.32
Bronchoscopy and bronchoalveolar lavage
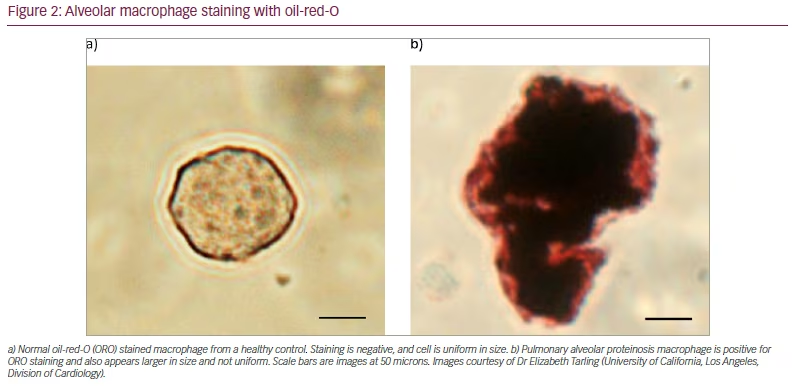
Bronchoscopy with bronchoalveolar lavage (BAL) is commonly performed to ascertain the presence of PAP. In patients with PAP, the BAL fluid appears grossly white to yellow in colour, with a milky consistency and the presence of sediments. This milky substance is related to the high lipoproteinaceous content of the amorphous material within the alveolar spaces.12 The fluid also contains acellular eosinophilic material, which is characteristically positive for periodic acid Schiff and negative for alcian blue.12 Foamy alveolar macrophages are also present and stain positive with oil-red-O (Figure 2).
Pulmonary function tests
Depending on the severity of the disease, spirometry is often normal in patients with PAP, although some patients demonstrate a restrictive physiology.21 The diffusion capacity of the lung for carbon monoxide (DLCO) is frequently reduced and has been inversely correlated with disease severity.2 Additionally, patients may have oxygen desaturation and an increase in alveolar-arterial oxygen (A-aDO2) gradient on arterial blood gas (ABG), indicating a reduction in gas exchange.21 However, ABG is rarely performed in the clinical setting due to patient discomfort.
Laboratory tests
The majority of laboratory studies are usually normal in patients with PAP. The most notable biomarker for patients with PAP is lactate dehydrogenase, which is elevated in up to 80% of patients.21 Other proteins, including surfactant protein A, surfactant protein B, surfactant protein D, and Krebs von de Lungen protein-6 are usually increased,33,34 and correlate with lung disease activity. These findings, however, are nonspecific and thus rarely used in the clinical setting. Additionally, chitinase-3-like protein 1 (YKL-40), a glycoprotein produced by neutrophils, macrophages, and synovial and malignant cells, has shown promise as a reliable biomarker for PAP, with elevated levels in the serum and BAL fluid of patients with PAP.35 In aPAP, neutralizing antibodies against GM-CSF are elevated in the serum and BAL fluid, and a positive test is 100% sensitive and specific for the diagnosis of aPAP.2,3,13,36 It is thus imperative to obtain GM-CSF autoantibody levels in any patients with suspected aPAP, prior to conducting more invasive studies and biopsies.
Diagnostic pathway
A positive GM-CSF autoantibody test confirms the diagnosis of aPAP, and is the preferred method for diagnosis in patients with symptoms and imaging that are compatible with aPAP. Patients with PAP who are found to have a negative GM-CSF autoantibody test are classified into secondary PAP, hereditary PAP, congenital PAP or unclassified PAP. Individuals who have a condition known to cause secondary PAP are diagnosed with secondary PAP. Patients with no known underlying causative conditions should be tested for serum GM-CSF levels and GM-CSF signalling.4
Results that show elevated serum GM-CSF levels and reduced or absent GM-CSF signalling, should prompt further testing for CSF2RA and CSF2RB mutations that identify hereditary PAP.37 If CSF2RA and CSF2RB mutations are absent, but the individual has a histology that is compatible with PAP, individuals are then diagnosed with unclassified PAP.4
If results indicate physiological levels of serum GM-CSF and GM-CSF signalling, tests should be performed for mutations in surfactant-related genes. If positive for these mutations, then individuals are diagnosed with congenital PAP. If negative, and histology is compatible with PAP, they are then diagnosed with unclassified PAP.4 Notably, diagnoses other than aPAP or secondary PAP remain rare.
Management
Some patients with PAP remain asymptomatic and do not require treatment at all. For those who develop progressive disease, treatment is focused on alleviating symptoms, preventing further disease progression, and improving quality of life. Currently, there is no cure for PAP, and specific treatment strategies depend on the underlying disease that is causing the PAP.3 Patients who progress to end-stage respiratory failure, typically due to pulmonary fibrosis, may require lung transplantation.3,38,39
Whole-lung lavage
WLL is considered the current standard of care for treatment of aPAP,40 and has been a cornerstone of PAP therapy since its introduction in the 1960s.12 In addition to aPAP, WLL can be used to treat some causes of secondary PAP, but not congenital PAP.3 WLL is an invasive procedure that requires general anaesthesia. A double-lumen endotracheal tube mechanically ventilates one lung, while the other is repeatedly filled with saline (up to 50 L per lung), to physically clear the surfactant sediment.3 The procedure is typically performed in two separate sessions, with one lung being treated per session.
WLL is typically pursued due to worsening respiratory symptoms that affect quality of life, and/or a decline in pulmonary function testing.3,39 Although it is widely used to improve symptoms, radiographic findings and oxygenation in patients,19,41–43 there are no standardized or specific indications for WLL.3,19,39 It remains highly operator-dependent and may involve concomitant thoracic percussion to enhance removal of protein, washing of the contralateral lung during the same procedure, and changing of positions during lavage.40,44,45 No prospective studies or randomized clinical trials have demonstrated the true efficacy of WLL. Retrospective data, however, show an increase in survival with the 5-year actuarial survival rate (standard deviation) from diagnosis being 94% (± 2%) in those who underwent WLL compared with 85% (± 5%) for those who did not undergo WLL.21 Interim results from a phase II trial have also shown that WLL used in combination with inhaled exogenous GM-CSF (described in further detail below) may be more effective than WLL alone.46
Complications of WLL include fever, hypoxemia, wheezing, acute respiratory distress syndrome, pneumonia, pneumothorax, pleural effusion, cardiac arrest, and rarely, death.3,43,47 The differences in technique, however, make it challenging to interpret this data clinically. Mortality may be underestimated, since the majority of data is derived from case reports.
Exogenous granulocyte–macrophage colony-stimulating factor
Subcutaneous granulocyte–macrophage colony-stimulating factor injections
Treatment of aPAP with exogenous GM-CSF was first attempted by Seymour et al. in 1996, via subcutaneous injection of recombinant human GM-CSF, and resulted in marked improvement in symptoms and arterial oxygenation.48 Several subsequent studies have reported improvements from the administration of subcutaneous GM-CSF.49 Three prospective studies used daily injections of GM-CSF at 5–9 μg/kg/day for 12 weeks, with a gradual increase in dosage to 18 μg/kg/day depending on the clinical response.50–52 Among these studies, treatment was effective in 43% of patients in the longest study (1 year), and as high as 75% of patients in the shortest study (12 weeks).51,52 Adverse reactions were generally minor, including oedema, erythema and pain at puncture site, fever, chills, nausea, vomiting, malaise, headache, fatigue, arthralgia and dyspnoea.40,49,50
Aerosolized granulocyte–macrophage colony-stimulating factor
Following concerns over the systemic toxicity of subcutaneous GM-CSF, subsequent trials have focused on the use of inhaled GM-CSF. Sheng et al. performed a meta-analysis to compare the delivery of GM-CSF via subcutaneous injection versus inhalation, which included 10 studies published between 2000 and 2016. Five of the included studies used subcutaneous GM-CSF therapy, and five used inhaled GM-CSF.49 While both routes were efficacious, inhaled GM-CSF led to a higher response rate than subcutaneous (89% versus 71%; p=0.023), with more improvements in alveolar oxygen partial pressure (PaO2) and A-aDO2 gradient.49 Additionally, inhaled GM-CSF delivery allows doses to be administered less frequently, reducing the cost of treatment.40
Two major multicentre, randomized, placebo-controlled clinical trials:,the PAGE trial and the IMPALA trial were completed to examine the efficacy of inhaled GM-CSF in aPAP.53,54 The PAGE trial examined patients with aPAP with mild-to-moderate disease, who were treated either with 125 μg of inhaled GM-CSF (sargamostim) twice daily for 7 days, every other week, or placebo for 24 weeks. The mean change in the A-aDO2 gradient was significantly higher in the GM-CSF group compared with the placebo group (-4.50 ± 9.03 mm Hg versus 0.17 ± 10.50 mm Hg; p=0.02); however, no other clinical benefits were demonstrated.53 In the IMPALA trial, 138 participants with aPAP were randomized into three groups: two groups received 300 μg of inhaled GM-CSF (molgramostim) either daily or intermittently (every other week), and one group received placebo. At 24 weeks, patients who received daily inhaled molgramostim had significant improvement in the mean change in A-aDO2 gradient from baseline and overall functional health status, compared with patients on placebo (A-aDO2: -12.8 mm Hg versus -6.6 mm Hg; estimated treatment difference, -6.2 mm Hg; p=0.03; SGRQ: -12.4 points versus -5.1 points; estimated treatment difference, -7.4 points; p=0.01).54 Additionally, there was greater improvement in multiple endpoints in the continuous inhaled GM-CSF group compared with the intermittent group. There were similar adverse events in all the groups except for chest pain, which was more frequent in the daily inhaled molgramostim group (22%).54 Both trials showed promising results but the findings were insufficient for regulatory approval.
Currently, the IMPALA-2 trial is actively enrolling patients with aPAP (ClinicalTrials.gov identifier: NCT04544293).55 It is a multicentre, randomized, placebo-controlled study evaluating the efficacy of 300 μg inhaled molgramostim daily versus placebo in patients with aPAP. The primary endpoint is the change in percent predicted DLCO from baseline at 24 weeks. Notably, although inhaled GM-CSF has not received any regulatory approval as of yet, molgramostim was granted Fast Track designation and Breakthrough Therapy designation in December 2019 by the United States Food and Drug Administration for treatment of aPAP, and the Promising Innovative Medicine (PIM) designation in August 2022 by the UK’s Medicines and Healthcare Products Regulatory Agency. PIM designation identifies molgramostim as a promising candidate to address unmet medical needs in the PAP disease space, and is an early indication that molgramostim could be eligible for the UK’s early access to medicines scheme. This is a programme that provides an opportunity for therapies in the UK to be used alongside later stages of the regulatory process.56–58
Rituximab
Rituximab is a monoclonal antibody against the B-lymphocyte antigen, CD20, and has been used to treat a variety of autoimmune disorders.3,59 There have been mixed results in regards to the efficacy of rituximab for aPAP. In 2011, an open-label, proof-of-concept phase II trial examined nine patients with aPAP who received two 1,000 mg intravenous doses of rituximab 15 days apart, and seven patients showed an improvement in PaO2 and A-aDO2 gradient, as well as in lung function and imaging scans.60 Treatment was well tolerated in patients with no major adverse events. Anti-GM-CSF autoantibody levels in the lung were reduced between the baseline and the 6-month timepoint, but serum levels during the same window were unchanged. A more recent retrospective study was performed in France with 13 patients.61 No patients showed improvement after 6 months of treatment, and four patients had a decrease in A-aDO2 gradient after 1 year. The study concluded that the data did not support rituximab as a second-line therapy, and that it should be used only for patients with refractory aPAP.
Plasmapheresis
Plasmapheresis treatment has rarely been used to decrease circulating levels of anti-GM-CSF antibodies. An initial case report showed a reduction in anti-GM-CSF autoantibody titre, as well as associated improvements in symptoms, oxygenation and imaging.62 Two subsequent case reports showed mixed results,63,64 and further studies are needed to elucidate whether plasmapheresis is an effective therapy for patients with aPAP.
Lung transplantation
Lung transplantation has been performed for patients with end-stage fibrosis, and paediatric patients. In one case report, an adult patient with refractory PAP showed recurrence of disease 3 years after bilateral lung transplantation.65 In the paediatric population, one small case series (n=3) showed no recurrence of disease 2 years after transplantation,66 while another case report showed evidence of recurrence.67
Prevention of secondary infections
Secondary infections cause significant morbidity in patients with PAP, therefore active vigilance for infectious complications, as well as routine vaccinations for influenza, COVID-19 and pneumococcus are recommended. Prophylaxis with trimethoprim/sulfamethoxazole may be considered for patients with significant disease.39
Other emerging therapeutic areas
Targeting lipid homeostasis
The accumulation of cholesterol in alveolar macrophages due to deficient GM-CSF signalling has been identified as a key defect driving the pathogenesis of PAP.11 Thus, treatment strategies targeting pulmonary lipid homeostasis have emerged as a novel therapeutic approach.
Oral statin therapy has been shown to reduce PAP disease and cholesterol accumulation in PAP mice models, by increasing cholesterol efflux in alveolar macrophages. In patients with aPAP, statin therapy was associated with significant clinical and radiographic improvement.11 Oral peroxisome proliferator-activated receptor gamma (PPAR-γ) agonists have also been shown to decrease cholesterol accumulation in GM-CSF deficient mice by activating PPAR-γ, a downstream transcription factor in the GM-CSF signalling pathway, which increases cholesterol clearance.68 An open-label, phase I/II clinical trial studying the safety and efficacy of oral pioglitazone in aPAP has recently been completed but results have yet to be published (ClinicalTrials.gov identifier: NCT03231033).3,69 These studies suggest that statins, and other drugs targeting cholesterol, may be novel therapeutic approaches to PAP. On-going lipidomic research of patients with PAP presents an opportunity to further understand the pathogenetic nature of alveolar cholesterol accumulation, and an avenue for related therapeutics.
Unmet needs
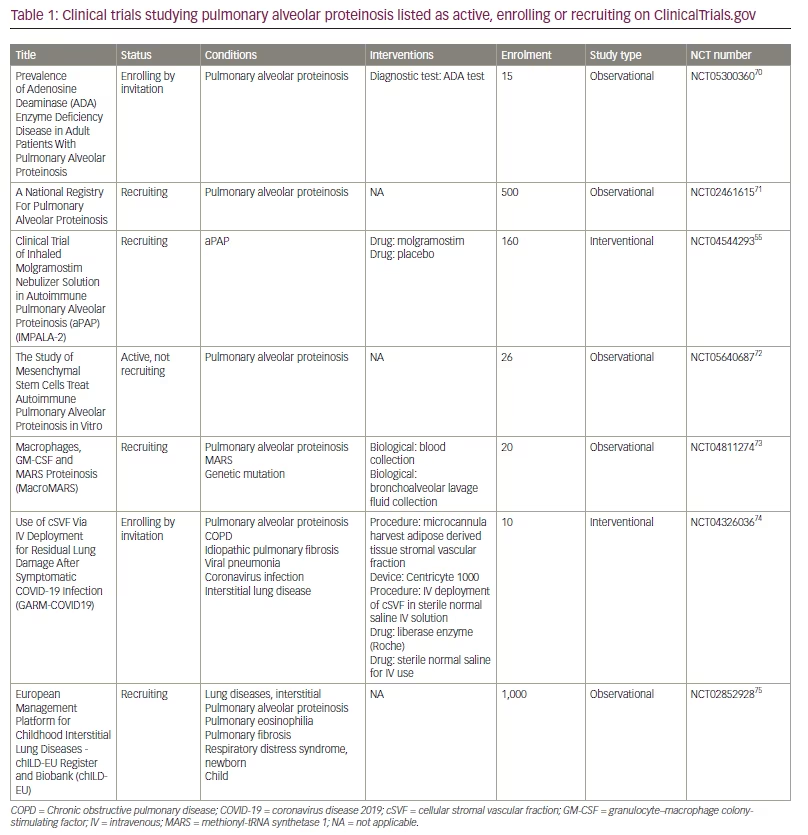
There is no known cure for PAP, and treatment of the disease is limited to symptom management, prevention of disease progression, and improvement in quality of life. WLL remains the gold-standard treatment but is an invasive procedure that is not standardized, and is highly operator-dependent.19 Inhaled recombinant GM-CSF has shown promise in recent clinical trials, but questions remain as to the appropriate dose, frequency, and optimal duration of treatment. More research is required to identify factors that may predict the response rate to inhaled GM-CSF in patients with aPAP, as some patients do not respond to treatment.39 While preliminary results have shown that WLL used in combination with inhaled GM-CSF may be more effective than WLL alone, no further clinical trials have investigated the combination of WLL and GM-CSF as an intervention for PAP.46 Case reports have also shown symptom improvement in patients with aPAP on oral statin therapy, but there has yet to be a clinical trial to assess the efficacy of statins. A full list of active, recruiting, and enrolling clinical trials related to PAP is shown in Table 1.55,70–75 Further research into the lipidome of patients with PAP presents an opportunity not only to elucidate pathogenetic mechanisms, but also to identify novel therapeutic targets and disease biomarkers. Such insights could indicate whether the alveolar lipidome alters in response to disease treatment or correlates with clinical profiles.76
Conclusion
Pulmonary alveolar proteinosis is a rare pulmonary syndrome in which surfactant accumulation in the alveoli leads to impaired gas exchange. In this article, the pathogenetic mechanisms of the three types of PAP (primary, secondary, and congenital) are reviewed, as well as the standard clinical course, relevant testing, and diagnostic pathways. Management of PAP is limited to alleviating symptoms and preventing disease progression, and WLL, an invasive procedure that requires general anaesthesia, remains the standard of care. Efficacy and safety data of alternate therapeutics are reviewed, including prior and current investigations in the treatment of PAP. Specifically, inhaled, exogenous GM-CSF has shown promise as a treatment for aPAP and is currently being investigated in the IMPALA-2 clinical trial. Treatment strategies targeting pulmonary lipid homeostasis have additionally emerged as a novel therapeutic approach and continue to be an area of active research. Continued investigation into the optimal treatment of the various forms of PAP, as well as further insight into pathogenetic mechanisms, remains key to improving the standard of care for patients in this rare disease space.