Duchenne muscular dystrophy (DMD), the most common and devastating type of muscular dystrophy,1 is characterised by the absence of the protein dystrophin, which causes premature muscle cell failure and leads to progressive muscle atrophy and loss. The condition is typically diagnosed at age 3–5 years when children start to show signs of physical disability including difficulty in walking. Patients experience progressive muscle weakness and become non-ambulant in early teenage years.2 From the age of 10–12 years, pulmonary function typically starts to decline3–5 and cardio-pulmonary complications are the major cause of morbidity and early mortality in DMD.6 Progressive pulmonary insufficiency requires the use of non-invasive and, in some circumstances, invasive ventilation, which, following the loss of ambulation, is the second irreversible disease milestone in DMD greatly impacting the patients’ quality of life. Although standard of care recommendations, including recommendations to monitor pulmonary function, have been proposed,2,6–8 overall compliance with these recommendations generally appears to be poor in real-world DMD care.9
DMD patients and their caregivers consider the preservation of pulmonary function, particularly maintaining effective cough and reducing the risk of airway infections, an important treatment goal,10 which at the current time represents a significant unmet need.
The aim of this review is to provide an update on advances in the understanding of DMD, its natural history and recent pharmacotherapies. We will summarize advances in the understanding of pulmonary function assessment in patients with DMD and the natural course of pulmonary function decline. We will also report clinically relevant thresholds of pulmonary morbidity in the context of standard of care recommendations. Finally, we will review the effects of glucocorticoids (GCs) on pulmonary function outcomes and present emerging evidence that the investigational drug idebenone can slow the loss of pulmonary function in DMD patients.
Assessment of pulmonary function in patients with Duchenne muscular dystrophy
Serial assessment of pulmonary function is a critical element of routine monitoring for patients with DMD, as it may enable early identification and treatment of pulmonary complications. In the absence of obstructive pulmonary disease, forced vital capacity (FVC, measured in litres), peak expiratory flow (PEF, measured in litres/minute), and forced expiratory volume in 1 second (FEV1, measured in litres) are well-established dynamic spirometry measures useful in the assessment of restrictive pulmonary function changes due to neuromuscular weakness. In DMD, both inspiratory and expiratory respiratory muscle weakness can be indicated by low peak inspiratory and expiratory flows.5,11 As pulmonary function is influenced by body growth and age, these measures are typically normalised relative to height and age12,13 and expressed as a percent of predicted (PEF%p, FVC%p, FEV1%p). As assessment of standing height in patients with DMD could be complicated by an inability to stand fully erect, scoliosis and joint contractures (particularly in non-ambulant patients), height can be estimated from ulna length measures.14,15 Although effort dependent, FVC%p and PEF%p can reliably be measured in school-age patients as shown by low within-subject coefficients of variation (CV) for successive measures.16
Natural course of pulmonary function decline in Duchenne muscular dystrophy
Two recent prospective studies investigated the natural course of pulmonary function loss in DMD. The Cooperative International Neuromuscular Research Group (CINRG) Duchenne natural history study (DNHS) is an ongoing, prospective international natural history study and currently represents the most comprehensive description of a longitudinal observational cohort of DMD subjects.4,17 Independently, prospective data in DMD patients were collected in the Neuromuscular Clinic at the Children’s Hospital of Philadelphia from 2005–2010 as part of an Institutional Review Board approved United Dystrophinopathy Project (UDP) cohort study.5
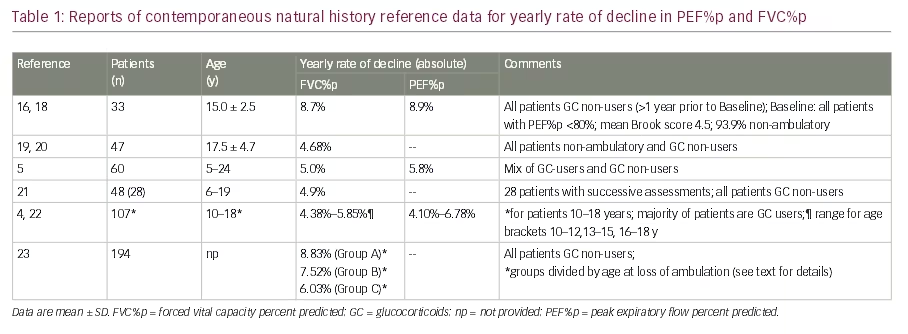
As Figure 1 from the CINRG-DNHS data set shows, flow-related PEF%p and volume-related FVC%p decline in a similar pattern, with decline in PEF%p slightly preceding the decline in FVC%p. At around 10–11 years of age, both parameters fall below the 80% predicted threshold, which is generally defined as the lower limit of normal and defines restrictive respiratory disease or low lung volume.7 From the 80% threshold, both PEF%p and FVC%p follow a co-linear decline, approaching a floor of approximately 20% of predicted from age 20 onwards.
This parallel and tightly correlated decline between FVC%p and PEF%p from early teenage years was independently reported from the data set collected at the Neuromuscular Clinic at the Children’s Hospital of Philadelphia.5
From this and other data collections, yearly rates of absolute decline in FVC%p and PEF%p can be extracted (Table 1). Although not directly comparable due to differences in age, GC-use status and other factors at time of assessment, these data indicate a yearly decline well above 5% absolute for both pulmonary function test (PFT) measures.
Correlation between pulmonary function and general disease stage in Duchenne muscular dystrophy
It would be of clinical relevance if loss of pulmonary function could be predicted from or at least correlated with the general disease stage of DMD. As seen from Figure 1, crossing the 80% of predicted threshold for PEF%p or FVC%p and subsequent decline generally coincides with the time DMD patients become non-ambulant during early teenage years.
This is supported by baseline data from the DELOS trial18 in 10–18-year-old DMD patients (n=64) where the majority of enrolled patients (92%) were already non-ambulant and at baseline had declined in pulmonary function to PEF%p of 53.8% (standard deviation [SD] 11.8) and FVC%p of 52.8% (SD 18.1).16,18 The predictive value of the age of loss of ambulation was more systematically analysed in a French database with 278 DMD patients.23 This study separated three groups of patients according to their age when they became non-ambulant (age at loss of ambulation – group A: before 8 years; group B: 8–<11 years; group C: 11–<16 years). Longitudinal analyses indicated that early loss of ambulation correlated with patients beginning to lose lung volume sooner, or having a peak vital capacity sooner (group A: 10.26 years; group B: 12.45 years; group C: 14.58 years).23 Moreover, the annual rate of decline in FVC%p was also largest in patients who became non-ambulant at a very young age compared to patients who became non-ambulant at an older age (Table 1).
Earlier work by Anne Connolly and co-workers of the Muscular Dystrophy Association DMD Clinical Research Network19,20 motivated attempts to correlate pulmonary function status with the patient’s score on the Brooke Upper Extremity Functional Scale.24 The Brooke scale scores upper extremity function from ‘1’ (patient able to abduct arms into a full circle until they touch above the head), to ‘6’ (hands cannot be raised to the mouth). For patients enrolled in the CINRG DNHS it could be shown that successive loss of upper extremity function is correlated with progressive loss of pulmonary function (Figure 2). Specifically, pulmonary function decline becomes apparent when patients reach Brooke score 2 or 3, which marks the transition of whether a patient can or cannot reach overhead. Of particular relevance is the transition from Brooke score 4 to score 5 (representing whether the patient still can raise the hand to the mouth or is losing this functional ability). A Brooke score of 5 appears to be an indicator that FVC%p has likely dropped below 50%, which is a clinically relevant threshold of pulmonary function loss (see below). These findings were independently confirmed by baseline data from the DELOS trial.18 Here, patients with Brooke score 1–4 on average had PEF%p, FVC%p and FEV1%p of 60–80% of predicted, whereas in patients who lost the ability to raise their hand to their mouth (Brooke score 5), mean values for PEF%p, FVC%p and FEV1%p dropped below 50%.16
In summary, recent data indicate that pulmonary function loss can be correlated with the general disease stage of the patient. The age of loss of
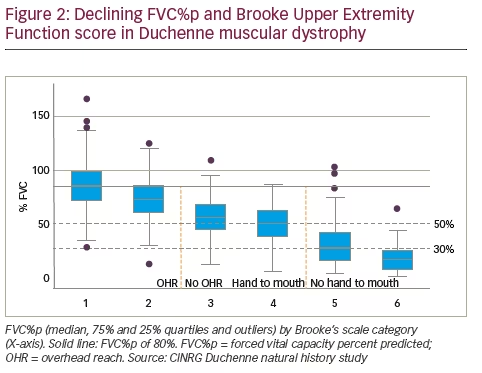
ambulation or the upper extremity function status may therefore provide readily available clinical assessments, which could indicate the need for more intense pulmonary function monitoring and clinical intervention.
Sleep-disordered breathing in children and adolescents with Duchenne muscular dystrophy
In DMD, progressive respiratory muscle weakness will eventually cause nocturnal hypoventilation and later daytime hypoventilation and the need for continuous mechanical ventilation. However, nocturnal hypoventilation is typically considered more broadly within the category of sleep-disordered breathing (SDB). SDB is most likely in rapid eye movement (REM) sleep, a period characterized of maximal muscle atonia.25,26 Clinical presentations of SDB can be restlessness in sleep, unrefreshing sleep or daytime fatigue.27 In DMD patients, SDB is often aggravated by pharyngeal muscle weakness leading to obstructive sleep apnoea (OSA), typically caused by snoring.27 The most comprehensive study published to date by Sawnani and co-workers28 indicates that OSA during REM sleep is the commonest form of SDB in younger DMD patients. Interestingly, this work identified high body mass index (BMI) as a risk factor for OSA, which is of relevance due to the risk of weight gain during GC treatment. Identifying daytime predictors of SDB is still an area of active clinical research in DMD.27 For example, occurrence
of hypopnoeas, nocturnal hypercapnic hypoventilation in REM sleep and continuous hypoventilation appeared to be correlated with a drop of inspiratory vital capacity.25,26 Another study29 established a drop in FEV1%p to <40% as a sensitive but not specific marker to predict sleep hypoventilation and hypoxic burden. Clearly, the identification of reliable and sensitive daytime predictors for SDB should constitute an area of future pulmonary function research in DMD.
Characterization of pulmonary morbidity in Duchenne muscular dystrophy
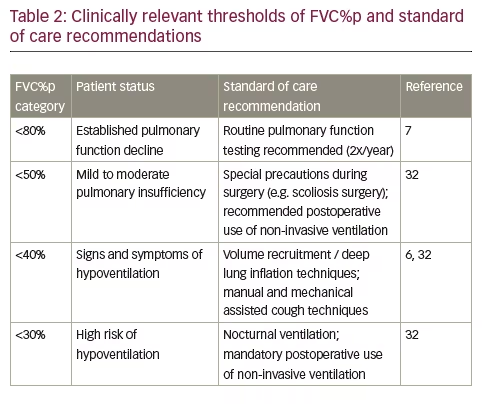
Although FVC%p and PEF%p follow a parallel trajectory of decline, as shown in Figure 1, decline in FVC or FVC%p is historically the best validated pulmonary function predictor of early morbidity and mortality in patients with DMD.7,30 As soon as patients are non-ambulant and FVC%p drops below 80%, regular (2x/year) consultation by a physician specializing in paediatric respiratory care is recommended.7 Once FVC%p drops below 50%, DMD patients may experience early clinical symptoms such as nocturnal hypoventilation, waking at night, morning headaches and problems with concentration and are considered at increased risk for post-procedural respiratory insufficiency when they undergo general anaesthesia or procedural sedation. The risk increases further once the FVC%p declines below 30%.31 Hence, the probabilities to present moderate pulmonary insufficiency is linked to FVC%p of <50% and severe pulmonary insufficiency in DMD is considered when patients have FVC%p <30%,6,23 and preoperative training in and postoperative use of noninvasive ventilation is recommended for patients with FVC%p <50% but is necessary for patients with FVC%p <30%.32 The risk of post-operative respiratory complications can be minimized with aggressive use of airway clearance and non-invasive ventilation as a bridge from invasive ventilation. It can be particularly helpful to introduce the assisted airway clearance strategies in the preoperative period if the patient does not already have an effective airway clearance regimen.
In addition, a drop in FVC%p below 40% is recommended as a threshold below which manual and mechanical assisted cough techniques are necessary to support effective airway clearance.6,32 Transition between these clinically relevant pulmonary function categories commands a change in patient care and management, as summarised in Table 2.
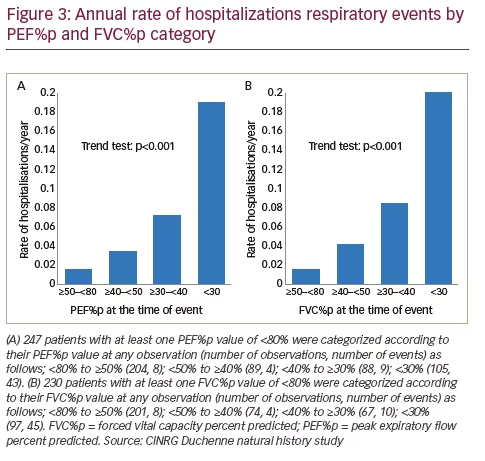
Like a drop in FVC%p, a decline in PEF%p is also directly correlated with bronchopulmonary complications, such as airway infections, and the risk of hospitalization for any respiratory reason, as shown from data obtained at the CINRG DNHS (Figure 3). Peak cough flow (PCF) is the peak flow generated during a cough and is used to objectively define airway clearance. Specifically, when the PCF falls below 160 l/min, the likelihood of successful extubation and airway clearance during an acute respiratory illness declines substantially, and patients with a PCF below 270 l/min are likely to fall below 160 l/min when viral illnesses occur and have problems with airway clearance.6,32–35 However, this risk can be managed with the use of assisted airway clearance therapy. Therefore, once PCF declines below 270 l/min it is recommended that patients be started on assisted airway clearance with the cough assist device.32,33 This is important because in the face of inadequate airway clearance, an otherwise benign upper respiratory tract infection can lead to lower respiratory tract infections and place the patient at risk of acute respiratory failure.
Clearly, the introduction of ventilatory support in routine clinical care for patients with DMD has considerably increased life expectancy36 and recent reports indicate that median age of survival in patients using ventilator support is presently around 27 years or above.37,38 Nevertheless, respiratory failure and associated complications are still a main cause for mortality even in ventilated patients39 and in patients using GCs.40
Glucocorticoids delay the onset but not the rate of pulmonary function loss in Duchenne muscular dystrophy
There is general agreement that ambulatory patients with DMD benefit from GC treatment,41 often started already in patients as young as ~5 years of age.2 The therapeutic objective of GC treatment is to slow the decline in muscle strength and stabilize pulmonary function. The use of GC has been shown to prolong ambulation by several years,42 but the majority of GC-using patients become non-ambulant by approximately 14 years of age. More recently, continued treatment after the patient becomes non- ambulant has also shown clinical benefit, such as reduction in the risk of progressive scoliosis, stabilization of pulmonary function and delay in the loss of upper limb function.4,43–47 The results of these and other clinical reports support the use of GCs, which have become the mainstay of DMD disease management.48–50 However, their chronic use in clinical practice has been hampered by significant adverse events and not all patients respond to GC to the same extent.
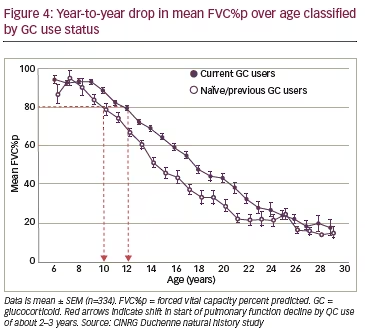
The effect of GC use on pulmonary function has been analysed from the CINRG DNHS.4,17 These data show that onset of pulmonary function loss is delayed by ~2–3 years in patients receiving GCs compared to patients not on GC therapy (Figure 4). However, further analyses of the CINRG dataset show that once pulmonary function decline is established (i.e. falling below 80% of predicted), the year-to-year drop in mean FCV%p by year of age on average is similar in all patients (i.e. those using GCs vs. GC-naïve or previously GC-treated patients) down to 20 years of age where the FVC%p of GC-naïve patients approaches a nadir. Importantly, despite the use of GC, pulmonary function decline continues linearly and unabated from the age of approximately 10 years through to the early twenties (Figure 4).
This is important in light of the well-described risks associated with chronic administration of GCs such as growth retardation, bone demineralization and increased fracture risk, obesity, insulin resistance and hypertension, among others.2 In the CINRG DNHS, 38% of DMD patients were either GCnaïve (24%) or had discontinued GC treatment (14%), with a clear trend to a higher proportion of GC non-users in the later post-ambulatory phase of the disease. In fact, for the patient group 10 years and older, 42% of DMD patients had never used GCs or discontinued their use because of side effects and tolerability limitations.4 Interestingly, a study of all-cause mortality showed that the number of deaths due to respiratory failure was not significantly influenced by the GC use status of patients.40
Therapeutic approach with idebenone to preserve pulmonary function in patients with Duchenne muscular dystrophy
There is an increasing body of literature indicating that dystrophin deficiency causes sarcolemmal fragility and intracellular Ca2+ dysregulation, which generally leads to mitochondrial dysfunction, a reduction in resting ATP levels, an increased production of cell-damaging reactive oxygen species (ROS) and ultimately mitochondrial damage (reviewed in 51). Mitochondrial activation via stimulation of Complex II- or Complex IIImediated electron flux has been explored as a therapeutic approach to overcome mitochondrial dysfunction in dystrophin-deficient muscle.11
Idebenone, a synthetic short-chain benzoquinone, has been reported to utilize and activate Complex I-independent metabolic pathways52–54 by the transfer of energy equivalents from the cytosol directly into the mitochondrial electron transport chain (ETC). Upon entering the cell, idebenone is efficiently reduced by the cytoplasmic enzyme NADH-quinone oxidoreductase 1 (NQO1) and the resulting reduced form of idebenone enters the mitochondria where it transfers its electrons to Complex III.52 Preclinical studies showed that idebenone had a cardio-protective effect and improved exercise performance in the dystrophin-deficient mdx mouse model of DMD.55
A proof of concept, randomized, placebo-controlled trial (DELPHI trial) in 21 DMD patients treated for 12 months provided initial clinical
evidence that idebenone might have the potential to slow down loss of pulmonary function.56,57
Based on the outcome of the DELPHI trial, the confirmatory phase III, randomized, placebo-controlled DELOS trial was conducted, which investigated the efficacy of idebenone on pulmonary function outcomes. The trial enrolled 64 DMD patients (age 10–18 years, 92% non-ambulatory at baseline) who discontinued GC use at least 12 months prior to study start. Eligible patients had to be in the pulmonary function decline stage, defined as PEF%p at baseline of <80%. Patients received idebenone (Raxone® [Santhera Pharmaceuticals, Liestal, Switzerland], three times 300 mg/day with meals) or matching placebo for 52 weeks. The trial met its primary endpoint and demonstrated a statistically significant and clinically relevant reduction in the loss of PEF%p.18
The observed treatment difference (between idebenone and placebo groups) for PEF%p for the change during the one-year follow-up period in the intention to treat (ITT) population was 6.27% (95% confidence interval [CI]: 0.61–11.93; p=0.031). This magnitude for the observed treatment difference is comparable to the reported natural rate of decline over one year (see Table 1), thus the treatment difference is clinically relevant for patients not taking GCs. The observed treatment difference for FVC%p of 3.27% (95% CI: -0.43–6.97; p=0.082) corresponds to approximately half the magnitude of the expected decline over one year in untreated patients.
The outcome of the primary endpoint was robust across pre-specified study populations and supported by findings for changes in PEF, FVC%p, FVC, FEV1%p and FEV1 (reviewed in 11) and inspiratory function.58 Interestingly, the treatment effect on PFTs was not influenced by the prior GC use status. Specifically, the estimated treatment effect for PEF%p and FVC%p was comparable in patients who never used GC and those who stopped GC use at least 12 month prior to study start.18 In addition, idebenone showed a larger effect size for PEF%p and FVC%p in the subgroup of patients ≤14 years of age than in the older patients (>14 years), suggesting that patients may derive a larger benefit from idebenone if treatment is initiated early.18
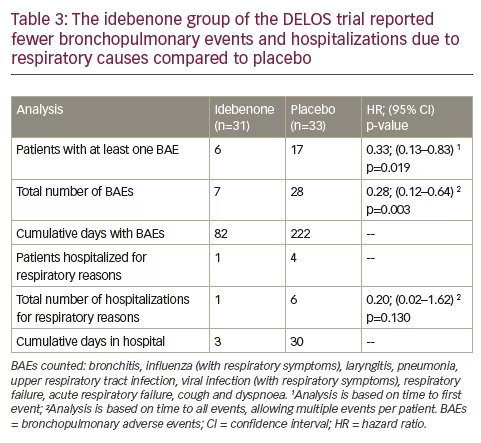
Additional analyses of the DELOS trial showed that more patients in the placebo group compared to the idebenone group experienced bronchopulmonary adverse events (BAEs).59 In the placebo group there were 17 patients reporting 28 BAEs, compared to six patients reporting seven BAEs in the idebenone group. The hazard ratios (HR) calculated for the time to first BAE (‘by patient’) (HR 0.33, p=0.019) and for time to any BAE allowing multiple events per patient (‘all BAEs’) (HR 0.28, p=0.003) indicated a clear idebenone treatment effect (Table 3). The overall duration of BAEs was 222 days (placebo) versus 82 days (idebenone). The higher frequency of BAEs in the placebo group was also reflected in the number of serious adverse events leading to hospital admissions due to respiratory causes. There were four patients in the placebo group hospitalized six times for a total of 30 days, compared to only one patient in the idebenone group hospitalized once for three days (Table 3). Since mortality from respiratory failure in DMD occurs during acute respiratory illness requiring hospitalization, these findings of reduced respiratory illnesses and hospitalizations is an extremely valuable outcome.
In addition, there was also a difference in the use of systemic antibiotics utilized for the treatment of BAEs. In the placebo group, 13 patients (39.4%) reported 17 episodes of antibiotic use compared to seven patients (22.6%) reporting eight episodes of antibiotic use in the idebenone group. Furthermore, patients in the placebo group used systemic antibiotics for longer (105 days) compared to patients in the idebenone group (65 days).
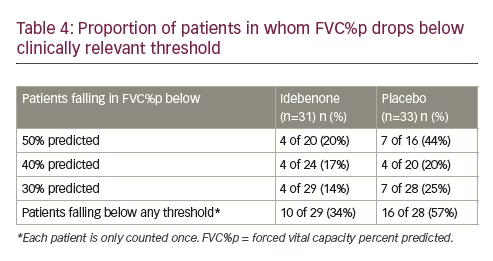
As mentioned in Table 2, a drop in FVC%p below clinically relevant thresholds commands changes in patient care and management. When the data from the DELOS trial were evaluated relative to the FVC%p thresholds described earlier, there was also benefit with idebenone treatment. In the idebenone group at study entry, 20 patients presented with FVC%p above 50% and in four (20.0%), the FVC%p fell below 50% during the study. In the placebo group, at study entry there were 16 patients with FVC%p above 50% at baseline and seven (44%) declined below 50% during the study (Table 4). Similarly, higher proportions of patients in the placebo group had FVC%p below the 40% and 30% thresholds respectively.
As shown, there was a higher proportion of patients in the placebo group falling below any clinically relevant threshold for FVC%p (idebenone: 34%; placebo: 57%), resulting in a HR of 0.51 (95% CI: 0.23–1.14; p=0.099) in favour of the idebenone treatment group.
In summary, emerging evidence supports the therapeutic potential of idebenone in slowing the decline in pulmonary function in DMD patients. Current evidence for efficacy is available for patients in their teenage years presenting with advanced disease (majority of non-ambulant patients) who were already in the pulmonary function decline stage. Available data consistently demonstrate a treatment benefit with idebenone in patients who were not using GC treatment and for whom there is currently no alternative treatment option. Patients eligible for idebenone treatment could include patients who previously were treated with GCs or in whom GC treatment is not desired, not tolerated or is contraindicated.
A new phase III, randomized, placebo-controlled trial (SIDEROS trial) is currently ongoing with the objective to further assess the efficacy of idebenone in DMD patients by examining the impact on pulmonary function in the phase of decline (FVC%p <80%) and in patients receiving concomitant GC therapy.60