Highlights
- Alpha 1 antitrypsin (AAT) deficiency (AATD) is a rare genetic risk factor that predisposes an individual to develop early-onset emphysema. It occurs most commonly in people of European heritage.
- Accurate diagnosis of severe AATD, including those cases associated with rare genotypes, is an important goal of clinical care as it facilitates early intervention. Despite this, AATD is widely under-recognised.
- Therapy with human plasma-derived AAT, which increases plasma AAT levels above the threshold considered protective for the lungs with the aim of slowing emphysema progression, is the only specific treatment of patients with AATD.
- The Randomized, placebo-controlled trial of augmentation therapy in Alpha1 Proteinase Inhibitor Deficiency (RAPID) study was the first randomised, placebo-controlled trial to demonstrate that emphysema progression is slowed by AAT treatment.
- RAPID, plus its open-label extension (OLE), currently comprises the largest completed clinical study of AAT therapy in patients with AATD and establishes the sustained efficacy and good tolerability of AAT treatment.
- Results from patients in the RAPID-OLE who received delayed AAT treatment following initial randomisation to placebo showed a greater deterioration in lung density than those who initiated AAT therapy earlier; they did not regain the lung tissue lost during the previous 24 months on placebo, indicating that the treatment modifies the course of the disease and supporting the importance of early intervention.
Alpha 1 antitrypsin (AAT) deficiency (AATD) is an under-recognised genetic risk factor that predisposes individuals to early-onset emphysema.1 This is due to insufficient concentrations of AAT, also known as alpha 1 proteinase inhibitor, which would normally protect elastin-containing tissues in the lung from damage by proteolytic enzymes.2,3 AAT is by far the most important antiprotease that needs to be present in adequate quantities to protect the lungs. The AAT alleles are autosomal co-dominant, i.e., the proteins that are coded by each allele present are produced in the liver and released into the blood stream. In normal genotypes with two Pi*M alleles, plasma levels are between 20–53 µM.4 Several mutations lead to reduced AAT serum levels. Patients with moderate AATD, e.g. with the Pi*SS, Pi*MZ and Pi*SZ genotypes, have serum AAT levels which are typically reduced to 40–60% of normal.4–6 This is thought to be sufficient to protect the lungs of non-smokers, while smoking individuals with moderate deficiency of AAT are more prone to develop lung disease than smoking Pi*MM individuals.7 Only a small proportion of individuals with a Pi*SZ genotype would be expected to develop emphysema, most often those with a history of smoking.8 Individuals with severe AATD have serum levels <15% of normal; in most cases this is associated with a Pi*ZZ genotype. The Z-AAT proteins polymerise and accumulate in the hepatocytes and are therefore unable to reach the blood stream, which leads to the deficiency. Also, due to injury caused by polymerised AAT in hepatocytes, liver disease such as cirrhosis and liver failure can occur in infants, children and adults.9 In some patients with AATD other rare clinical manifestations such as panniculitis and C-ANCA-positive vasculitis (i.e., Wegener’s granulomatosis) have been observed.9
Cessation of smoking is recommended in all individuals diagnosed with AATD, and approaches such as inhaler use, pulmonary rehabilitation, lung volume reduction and lung transplantation may be considered in some patients.3,10 However, in patients with emphysema due to severe AATD, infusion of human AAT is currently the only specific treatment for slowing the progression of emphysema associated with this genetic disease. The aim of this article is to describe the prevalence of AATD and the diagnostic strategies in Europe. For the different brands of AAT that are currently available, a brief overview of the product characteristics will be provided, and the clinical evidence available to demonstrate the disease-modifying effects of AAT therapy in AATD-related emphysema will be given.
Epidemiology
The most common deficiency-related alleles are Pi*Z and Pi*S which occur most commonly in people of European heritage (i.e., in Europe and North America), although they do occur at lower frequencies in all other racial subgroups worldwide.11 In Europe, the northern and western countries have the highest prevalence of the PI*Z allele, at a mean allele frequency of 0.0153. This gradually decreases throughout the rest of Europe from north to south, with the lowest prevalence in eastern Europe at 0.0092. The frequency of the PI*S allele is the highest in southern Europe (0.0564), decreasing in northern Europe (0.0176) (Figure 1).11–13 Accordingly, the resulting prevalence estimates for severe AATD Pi*ZZ vary considerably and can be as high as 1 in 1,668 inhabitants in Sweden, and 1 in 6,054 inhabitants in France, or 1 in 3,708 in Italy.11 AATD is therefore one of the most prevalent rare genetic conditions in Europe and North America.14
Diagnosis of alpha 1 antitrypsin deficiency
Diagnosis of AATD usually involves a combination of measurement of serum AAT levels, phenotyping using isoelectric focusing electrophoresis, and allele-specific genotyping to identify the most prevalent deficiencies (Z and S, sometimes additionally F and I).15 This latter technique requires mutation specific single nucleotide polymorphisms. However, these techniques are unable to detect other rare variants, which can only be detected by extended techniques such as whole gene sequencing, often not routinely available.16 Recently, next-generation sequencing genotyping is more frequently being used in genetic diagnostics and can provide additional information on regulator sequences of non-coding DNA.17
Accurate diagnosis of severe AATD, including those cases associated with rare genotypes, should be an important goal of clinical care as it enables early intervention, such as implementation of smoking cessation strategies, occupational counselling or drug treatments.15 In 1997, the World Health Organization recommended testing of all patients with symptomatic chronic obstructive pulmonary disease (COPD),18 and all subsequent guidelines and recommendations reinforce the need for testing of all adults with symptomatic COPD once in their lives.3,10,19 In addition, testing is recommended by the American Thoracic Society (ATS) and European Respiratory Society (ERS) for the following: all nonresponsive asthmatic adults and adolescents, all patients with cryptogenic cirrhosis or liver disease, granulomatosis with polyangitis, bronchiectasis of unknown aetiology, panniculitis, and first-degree relatives of patients with known AATD.3 The new ERS statement on AATD has outlined an extended algorithm for family screening of individuals diagnosed with severe AATD which includes first-degree relatives and spouses of affected individuals.10 Population screening or screening of healthy newborns or adolescents is not recommended.3


Despite dissemination of these guidelines, AATD is widely under-recognised, with less than 10% of expected individuals with severe AATD being identified.20–22 Lack of awareness about the disease and knowledge about testing methods may be one reason for under-diagnosis.23 Diagnosis is also hampered by the fact that the reported symptoms of AATD are largely identical to those of other obstructive lung diseases such as COPD. Non-smoking patients with a Pi*ZZ genotype, who may present with the disease decades later than Pi*ZZ patients who smoke, may be particularly prone to a misdiagnosis of COPD. The testing of all COPD patients is therefore one of the most important measures to detect AATD as outlined by the screening recommendations. Nevertheless, many patients with AATD experience a long delay – and often a frustratingly high number of physicians are consulted – between the occurrence of first symptoms and a diagnosis being made, during which time many patients will have experienced a major decline in their lung function.22–25 Other reasons for a delayed or missed diagnosis could include a tendency to only test younger patients presenting with lung disease and a misconception that existing treatments are inadequate.23,26 Other individuals may not be diagnosed because they don’t develop any overt lung or liver disease. AATD is a risk factor predisposing individuals to develop emphysema but an unknown percentage of individuals with severe AATD, particularly non-smokers, will remain undiagnosed because of the lack of symptoms.27
Treatment with alpha 1 antitrypsin
Intravenous (IV) therapy with AAT has been shown to be safe and well tolerated in patients with AATD-associated emphysema. Weekly IV infusions of 60 mg/kg body weight of AAT increase plasma levels beyond the protective threshold of 11 µM (57 mg/dl), protecting lung elastin and alveolar walls from destruction by uninhibited neutrophil elastase.28 Several AAT products are marketed and availability depends on the regulatory status of the respective country (Table 1). This review will focus on the products available in Europe: Respreeza® (CSL Behring, King of Prussia, PA, USA; marketed as Zemaira® in the US), Prolastin® (Grifols Germany GmbH, Frankfurt, Germany), and Alfalastin® (LFB Biomedicaments, Les Ulis, France).
Therapy with AAT is recommended by the ATS/ERS guidelines for patients with severe AATD and forced expiratory volume per second (FEV1) of 30–65%,3 although more recent consensus is to also consider treatment outside this range on a case-by-case basis.19 In patients with AATD-related lung disease and an FEV1 >65%, Alpha-1 Foundation practice guidelines recommend discussion with an individual patient on the potential benefits of therapy.19 A personalised approach to therapy should take into account the risk of continued rapid or long-term disease progression when identifying those patients for whom augmentation treatment is appropriate and those for whom the avoidance of risk factors may be enough to stabilise their condition.29 However, several years of follow-up of patients is required in order to detect significant changes in FEV1 and other measures of lung function, and it should be noted that should loss of lung tissue occur during this time due to a delay in treatment, this loss is irreversible.30
However, the high cost of augmentation therapy makes it particularly important that treatment is not given unnecessarily. An early cost-effectiveness analysis suggested that the annual cost of augmentation therapy would have to be reduced approximately tenfold in order to be considered a lifelong costeffective therapy,31 though morerecent analyses that also took into account the costs of potential lung transplantation have indicated that the cost per year of life gained with AAT therapy compares favourably with that of other accepted interventions, such as use of simvastatin for primary prevention of coronary artery disease, and breast cancer screening, particularly in non-smokers.32 If augmentation therapy is indicated, then there are few comparative trials to provide guidance to the physician as to which AAT agent to prescribe. Those performed to date have shown equivalence between AAT therapies on primary outcomes (generally serum or plasma AAT levels).10 With that in mind, the physician is left to make treatment decisions based on the agents available in their region and according to their own reading of the evidence presented for each agent in published guidelines.3,10
The published recommendations for management of AATD are based on a series of long-term follow-up lung function data from registries, and more recently, emphysema progression in randomised placebo-controlled trials. The relative rarity of AATD and the slow progression of the disease means that performing randomised clinical trials sufficiently powered to evaluate decline in lung function in terms of FEV1 is largely impractical;33 although, a meta-analysis carried out in 2009 including four studies of Prolastin and one study of Alfalastin was able to demonstrate a slowing of decline in FEV1 with treatment, and suggested that treatment efficacy was highest in patients with moderate lung function impairment.34 Alternative biochemical measures, such as serum or plasma AAT levels, have been used to demonstrate a treatment effect in lieu of clinical outcomes.29 However, the lack of studies showing an effect on clinical outcomes such as mortality, frequency of exacerbations and impact on quality of life, combined with the high cost of treatment, has led to some health authorities refusing to reimburse AAT augmentation.29,34
More recently, measurement of lung density by computed tomography (CT) has shown promise as a specific and sensitive marker of the progression of emphysema, which can reveal changes not detectable through spirometry.29 With the availability of quantitative CT scans to assess emphysema progression, this new tool rendered randomised clinical trials in AATD feasible for the first time. It was initially introduced in a study from Denmark and the Netherlands, in which 58 patients were randomised to receive AAT (Alfalastin) 250 mg/kg or albumin (625 mg/kg; placebo) infusions at 4-week intervals for at least 3 years. Mean ± standard error (SE) loss of lung tissue was 1.5 ± 0.41 g/L/year for AAT versus 2.6 ± 0.41 g/L/year for placebo (treatment difference 1.07 ± 0.58; p=0.07).35 In the Alpha-1-antitrypsin (AAT) to treat emphysema in AAT-deficient patients (EXACTLE) study, 77 patients were randomised to receive weekly infusions of AAT (Prolastin) 60 mg/kg or placebo for 2–2.5 years. At 30 months, mean ± SE change in lung tissue was -3.25 ± 4.5 in the AAT group versus -4.71 ± 4.51 in the placebo group (treatment difference 0.857; 95% confidence interval [CI] -0.065–1.778; p=0.068).36 In a post hoc analysis, data from 119 patients from these two studies were pooled to increase statistical power. Mean change in lung density from baseline to last CT scan was -4.082 g/L for AAT and -6.379 g/L for placebo with a treatment difference of 2.297 (95% CI, 0.669–3.926; p=0.006).37
The Randomized, placebo-controlled trial of augmentation therapy in alpha-1 proteinase inhibitor deficiency (RAPID) study was the first randomised, placebo-controlled trial powered to substantiate that emphysema progression is slowed by AAT treatment.38 To evaluate the clinical efficacy of AAT, the RAPID randomised controlled trial (RCT) (March 2006 to November 2010) used the primary outcome variable of annual rate of decrease in lung density. This was calculated as the change in the 15th percentile lung density (PD15) measured by CT. This technique, known as CT densitometry, allows the non-invasive quantification of the density of lung tissue through the analysis of CT images of the thorax.39 Any increase in the amount of emphysematous lung will increase the amount of air, resulting in lower lung densities. Consequently, any therapy-related preservation of lung tissue will result in a less pronounced decline in PD15. In the RAPID trial, 93 patients were randomly allocated to AAT IV 60 mg/kg per week (Respreeza/Zemaira) and 87 to placebo for 24 months.38 The annual rate of lung density loss at total lung capacity was significantly less among patients in the AAT group versus placebo (treatment difference 0.74 g/L/year; 95% CI 0.06–1.42; p=0.03). This corresponds to a reduction of 34% in the annual rate of lung density decline with AAT treatment versus placebo. Findings from the RAPID study support data from two previous studies that demonstrated a trend toward a slowed decline in lung density, assessed by whole lung CT scan reported as PD15, in response to treatment with human AAT (Alfalastin and Prolastin) in patients with AATD of the PI*ZZ phenotype.35,36
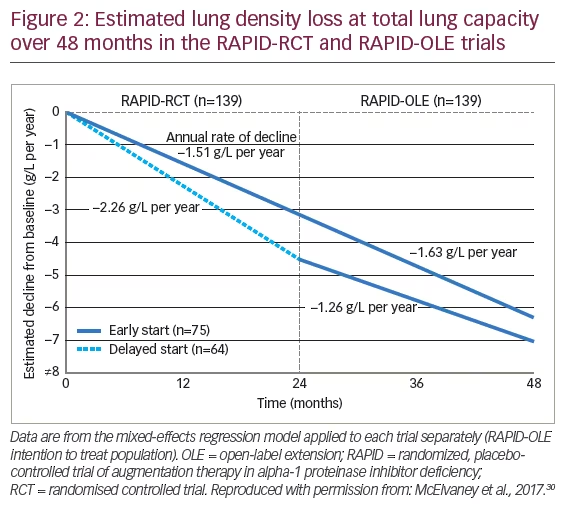
After completion of RAPID-RCT, 99% (140 out of 141 eligible patients) chose to enter a 24-month RAPID open-label extension (OLE) trial in which all patients received AAT treatment.30 Of these patients, 76 comprised the early-start group (defined as having received AAT in RAPID-RCT and who remained on AAT treatment) and 64 the delayed-start group (i.e., those who received placebo in RAPID-RCT and were then switched to open-label AAT). In the subsequent 24 months of RAPID-OLE, the rate of lung density loss was reduced in delayed-start patients and there was a significant difference in the slope of the first 2 years compared with the following 2 years when these patients had received AAT treatment. In contrast, the rate of lung density loss in early-start patients remained unchanged, indicating a consistent treatment effect (Figure 2). Thus, the slow decline observed in the early-start group was maintained over 48 months whereas the more rapid decline observed in the first 24 months in the delayed-start group was reduced to similar levels to the early-start group in the second year of the study once they had switched to active treatment. The RAPID-OLE highlighted the value of early treatment and the disease-modifying effect of AAT treatment since the patients in the delayed-start group did not regain the lung tissue lost during the previous 24 months when they were receiving placebo. In terms of safety profile, the RAPID study showed that AAT treatment was generally well tolerated with a similar tolerability profile to that of placebo. Treatment-emergent adverse events (TEAEs) were similar between AAT (1,298 TEAEs occurring in 92 [99%] patients) and placebo (1,068 occurring in 86 [99%] patients).38 According to the findings of an integrated safety analysis of six clinical trials (including RAPID and RAPID-OLE), adverse events occurred at a similar rate between 221 patients receiving AAT therapy and 87 receiving placebo, with the most common TEAEs being headache and dizziness.40
Disease modifying effect of alpha 1 antitrypsin therapy
Broadly speaking, disease modification is considered to be a sustained change in a disease state that occurs following therapeutic intervention.43 Thus, disease-modifying therapies should act on the pathological and pathophysiological mechanisms underlying the disease rather than the disease’s clinical or symptomatic effects. To date, RAPID-RCT and RAPID-OLE comprise the largest completed clinical study of AAT therapy in AATD patients and the only trial specifically designed to investigate the disease-modifying effect of treatment. The randomised start study design, in which patients were randomised to either an active-active or placebo-active treatment sequence, combined with the highly sensitive outcome measures used to assess efficacy, enabled the disease-modifying effect of AAT therapy to be identified. Importantly, patients who delayed treatment in this study (i.e., who switched from placebo to active treatment after they completed year 2) experienced a greater deterioration in lung density during the first 2 years than those who initiated AAT therapy earlier, emphasising the importance of early treatment. The mechanism of action of AAT is consistent with the disease-modifying effect in terms of reducing the rate of lung density loss: AAT preserves lung function by protecting the lung tissue and alveoli from degradation.44 In addition, although the original trial was not powered to show such correlations, in the RAPID trial programme study moderate-to-weak and significant 4-year correlations were observed between change in lung density loss at total lung capacity and spirometry values; overall correlation coefficient (r) was 0.338 for % predicted FEV1 (p=0.0002) and 0.286 for FEV1 (p=0.0017).30 Similarly, in the EXACTLE study, a statistically significant relationship was observed between the progression of CT densitometry and the rate of decline in FEV1 in patients treated with Prolastin.36 The slopes of CT lung density of patients in RAPID starting at baseline were extrapolated into the future. A cut-off of about 20 g/L was used when patients in RAPID either received a lung transplant or withdrew or died because of respiratory failure. Based on the different lung density loss between active and placebo this post hoc analysis estimated a prolongation of life by 5.6 years.30
Composition of human alpha 1 antitrypsin
Although the starting product in the manufacture of AAT is human plasma in all cases, the production process for each product differs, impacting the product characteristics. During the manufacturing process all products are purified and undergo several virus inactivation steps.
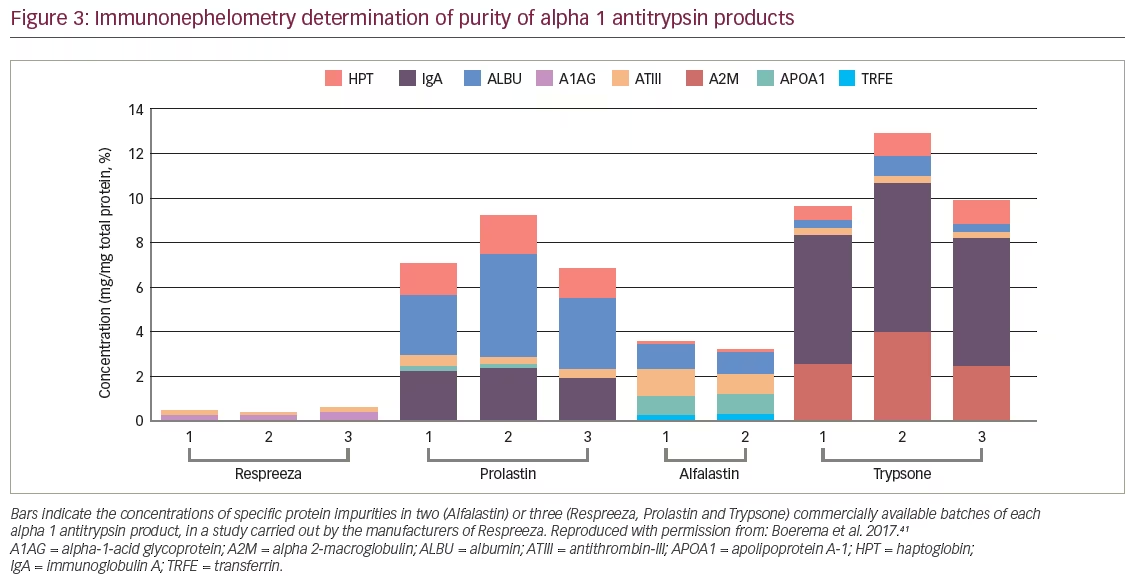
A recent study compared the four different AAT products available in Europe at the time of analysis (Respreeza, Prolastin, Alfalastin, and Trypsone® [Grifols, Los Angeles, CA, USA]) in terms of function, purity, structure and chemical modifications.41 Active AAT inhibits neutrophil elastase in a 1:1 fashion but the active protein is easily inactivated e.g., during the production process. As a result, all AAT products contain a mixture of active and inactive AAT as well as other plasma proteins, but their ratios differ. In comparison, Respreeza was found to have the highest level of functionally active AAT protein per volume of infusion-ready product (48.8 mg/mL); Alfalastin having the next highest level at 26.6 mg/mL. As AAT is dosed according to the amount of active protein, the high concentration of active protein per mL leads to a lower total infusion volume and shorter infusion time for Respreeza. Another parameter assessing product quality is the specific activity, which is defined as the ratio of functional protein to total protein. If all protein in the vial were active, the specific activity would be 1. Comparing the four products, Respreeza had the highest specific activity (0.862 mg active AAT per mg total protein, versus 0.775, 0.670 and 0.638 mg for Trypsone, Alfalastin and Prolastin, respectively). The remaining protein is either inactive AAT, which has been altered by the production process, or remaining plasma proteins. Impurities with other plasma proteins such as immunoglobulin A can be a cause of side effects. Compared with the other brands, Respreeza contained the fewest impurities and those were present in low amounts (Figure 3).41 The AATD products tested differ in purity, concentration, and chemical variation. These findings and the possible significance to the patient warrant further investigation.
The production process includes a number of highly effective pathogen inactivation steps. For Respreeza these are cold ethanol fractionation, pasteurisation and nanofiltration; their effectiveness has recently been assessed in detail.42 The methods were effective at removing all tested viruses below the limit of detection; in addition, cold ethanol fractionation enabled substantial reduction of prions. These findings confirm that the manufacturing process of Respreeza/Zemaira results in the production of a stable and pure product.
Impact of the European approval for Respreeza on the treatment landscape
The EU approval of Respreeza has expanded the number of countries in Europe in which AAT therapy is currently available (Figure 4). In the EU, Respreeza is indicated for maintenance treatment to slow the progression of emphysema in adults with documented severe AATD (e.g., genotypes Pi*ZZ, Pi*Z[null], Pi*[null,null] and Pi*SZ). Patients are to be under optimal pharmacologic and non-pharmacologic treatment and show evidence of progressive lung disease as evaluated by a healthcare professional experienced in the treatment of AATD.45 Respreeza is the only second-generation AAT inhibitor currently available in the EU although first-generation AAT inhibitors such as Alfalastin and Prolastin are available in some EU countries with a national licence.
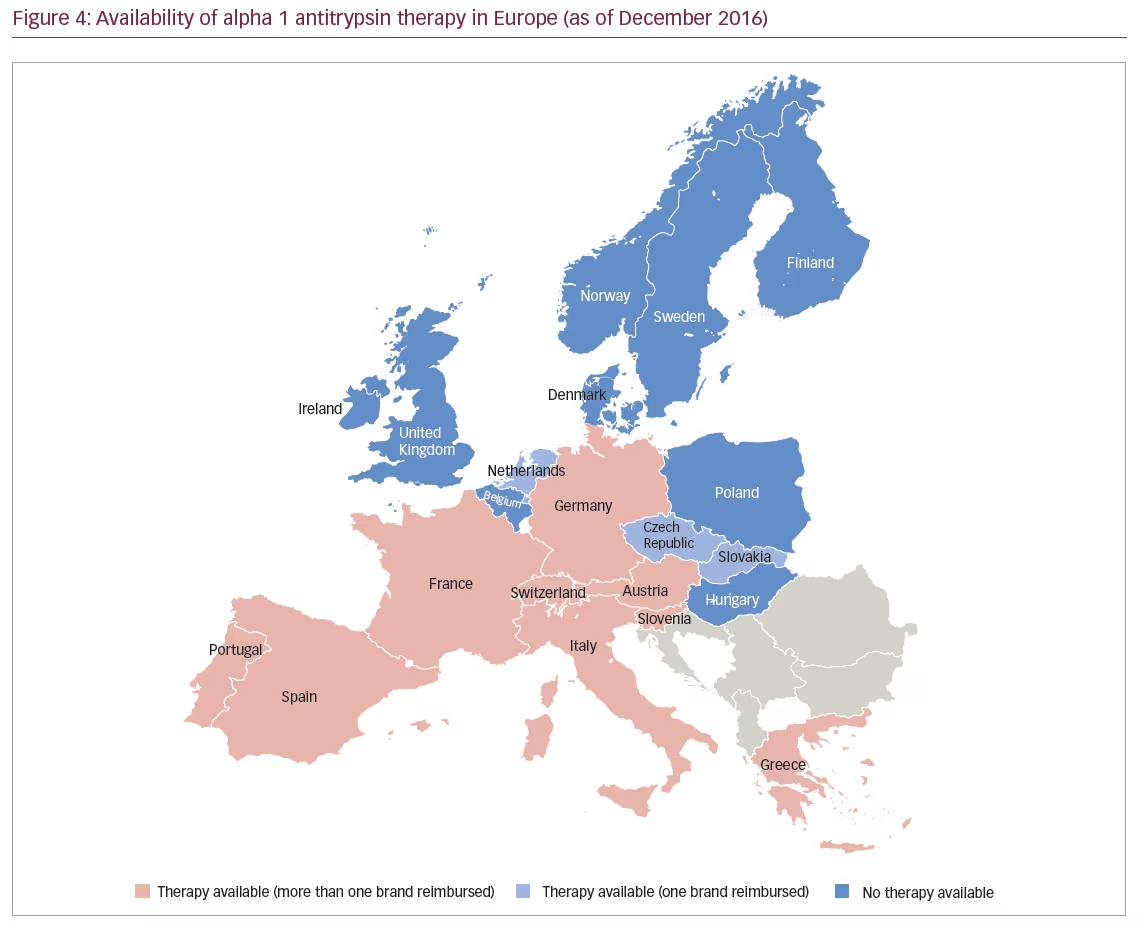
Unlike Prolastin, the use of which is restricted in the EU to patients with FEV1 35–60%, Respreeza has no FEV1 restriction.45 This is in accordance with new recommendations, for example the Alpha-1 Foundation practice guidelines, which recommend in AATD patients with an FEV1 >65% that physicians discuss with individual patients the potential benefits of therapy.19
Conclusions
AATD is substantially underdiagnosed, and is considered one of the most prevalent inherited risk factors among people of European ethnicity.46 The RAPID-OLE study confirms the findings of the RAPID trial, establishing the sustained efficacy for AAT treatment (Respreeza/Zemaira) and suggesting that slower lung density loss rates are achieved irrespective of the time at which treatment commenced.30 Furthermore, RAPID-OLE demonstrated that, between months 24–48, patients from the delayed-start group had similar rates of decline to the early start group and both were comparable with the AAT group during months 0–24. However, the delayed-start group experienced a greater deterioration in lung density than those who initiated AAT therapy earlier and they did not regain the lung tissue lost during the previous 24 months on placebo, highlighting the importance of early intervention.30 The mechanism of action of Respreeza is consistent with its disease-modifying effect: the therapy preserves lung function by preventing degradation of lung parenchymal tissue and alveoli by neutrophil elastase.44 In general, treatment with AAT is well tolerated.36,38 In RAPID-RCT, the incidence of TEAEs occurring with Respreeza was similar to that observed with placebo,38 a finding confirmed by an integrated safety analysis of six clinical trials of 221 patients receiving Respreeza.40 Furthermore, the RAPID trial was the first randomised, placebo-controlled study to show that purified AAT can significantly reduce the progression of emphysema, as assessed by measurement of lung density. Early introduction of treatment in patients with severe emphysema-related AATD may delay the time to death, lung transplantation or respiratory failure.47 Further studies are required to assess long-term outcomes.