Case report
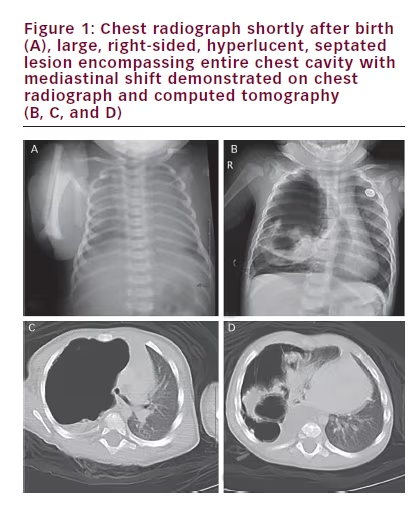
An eight-month-old girl presents with mild, intermittent tachypnea of several months duration. The girl’s past medical history was unremarkable except for a short neonatal intensive care unit (NICU) course secondary to respiratory distress following a term, vaginal delivery. She was meeting growth and developmental milestones. A chest radiograph taken while in the NICU was unremarkable (see Figure 1A). The repeat chest radiograph and subsequent computed tomography (CT) with contrast performed at an outside institution revealed a large, right-sided, septated cystic lesion causing mediastinal compression (see Figures 1B, 1C and 1D). Despite the use of intravenous (IV) contrast, the origin of the blood supply for the mass was difficult to ascertain with certainty on the CT images.
She was sebsequently transferred to a tertiary children’s hospital (Kosair Children’s Hospital, Louisville, KY) where she underwent evaluation by the paediatric pulmonary service as well as by paediatric surgery. There was initial concern that perhaps an endobronchial lesion or other airway anomaly had formed the cyst through a ball-valve mechanism. A congenital pulmonary airway malformation (CPAM) was another consideration but felt to be less likely given her normal chest radiograph at birth. To help exclude an endobronchial lesion, flexible bronchoscopy was undertaken. This procedure, however, was unrevealing of a cause, demonstrating normal airway anatomy apart from evidence of external compression of the right mainstem bronchus. The patient then proceeded to undergo exploratory thoracotomy and resection of the mass. She was monitored post-operatively in the paediatric intensive care unit (PICU) and overall had an uneventful course. Post-resection imagery showed complete re-expansion of the right lung. The intermittent tachypnea was no longer present once she recovered from surgery.
Operatively, the mass was found to be mostly free from the lung with small attachments at the superior portion of the mass anteriorly, around the middle and upper lobe. The mass was not intralobar and a number of small vessels were present and ligated in the posterior gutter and elsewhere. It was felt intraoperatively that the blood supply was entirely systemic as no pulmonary circulation could be identified. The mass was also noted to be grossly devoid of bronchial components and speculated to be air filled via accessory connections to the lung.
Grossly, the mass was brown-red and fully encapsulated by a shiny smooth pleural surface measuring 7 x 7 x 1.5 cm. The mass did not share a vascular supply with the lung, making it extralobar, which is unusual for a PPB. Sectioning showed multiple areas of dilated and tortuousappearing tissue grossly consistent with dilated vessels or cysts with haemorrhage. Microscopic examination showed a multicystic lesion
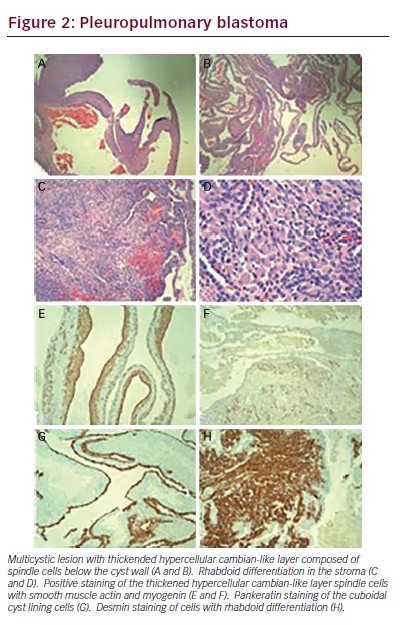
arising in a background of normal lung with bronchioles. The cysts exhibited a very cellular and primitive cyst wall that formed a cambiantype layer in areas with an unremarkable cuboidal cell lining (see Figure 2A and B). There were areas in the lung tissue that showed mature rhabdoid- type cells with abundant eosinophilic cytoplasm (see Figure 2C and D). There were occasional nodules of the immature spindled cells present in the cyst wall but no diffusely solid areas. The immature stroma showed strong cytoplasmic staining for desmin and smooth muscle actin and patchy nuclear staining for myogenin (see Figure 2E and F). The cuboidal lining cells were positive for Pankeratin AE1/ AE3 (see Figure 2G). The areas of rhabdoid-type cells showed diffuse strong cytoplasmic staining with desmin (see Figure 2H). PHH3 staining showed greater than 20 mitoses per 10 high power fields (not shown). These findings are all features of a type I pleuropulmonary blastoma (PPB), apparently arising from an extralobar sequestration. Only one other case of a PPB associated with an extralobar sequestration was identified during our literature search.1
Our patient was followed closely by our oncology colleagues who performed an abdominal and pelvic CT to evaluate for other lesions or masses, such as cystic nephromas, which was negative. The family was counselled on multiple occasions about the utility of DICER1 mutation analysis, however they did not agree to testing while being followed at our centre. The child had no other siblings and there was a strong family history of tumours with lung and colon cancer on the paternal side and throat and lung cancer on the maternal side. It is unclear if any of these tumours may also be linked to DICER1 mutations.
Discussion
Cystic lung lesions in paediatric patients are often congenital in nature. Most commonly they represent CPAMs, formally known as congenital cystic adenomatoid malformations, but on occasion can represent more ominous pathologies.2
The main differential diagnosis of this lesion was a CPAM type 4 (cystic) and the mixed cyst-solid and solid (types II and III) PPB. Pathologically, CPAMs do not exhibit the hypercellular cambian-type spindled cell layer under the cyst wall, do not exhibit positive smooth and skeletal muscle markers in those cells and would not be expected to show the prominent mitotic activity present in our specimen. The differential between types I, II, and III PPBs is based purely on the degree and amounts of solid and cystic tissue and requires generous sampling of the tumour, which was performed.
First described in 1988 as its own unique entity, PPBs represent an aggressive neoplasm often associated with DICER1 mutations. These mutations may be germline mutations or may arise sporadically, sometimes confined only to the tumour. DICER1 mutations result in dysregulation of microRNA, which provoke a cascade that can lead to oncogenic insults.3 Therefore, DICER1 mutations often result in a strong family history of unusual tumours. In addition to PPBs, patients with DICER1 mutations may be at heightened risk of a host of other tumours (both benign and malignant), including cystic nephroma, intestinal polyps, Sertoli-Leydig tumours, nasal chondromesenchymal hamartoma, ciliary body medulloepithelioma, Wilms tumour and rhabdomyosarcoma.4,5 As a result, DICER1 mutation analysis is important to help identify other at-risk family members and all families should be counselled on the benefits of undergoing genetic testing. PPB patients should also undergo a screening renal ultrasound or other appropriate imaging to evaluate for cystic nephromas and all patients and their families should be educated about their risk of future tumours. PPBs can present insidiously: they have been reported to commonly mimic more common pathologies with non-specific presenting symptoms, such as respiratory distress, cough and chest pain.6 In some cases, they may present as a recurrent pneumothorax7 or be misdiagnosed as CPAMs.
Early identification is crucial because while type I lesions can almost always be entirely resected and have an excellent overall prognosis, they can quickly progress to type II and type III lesions if untreated, which are far more invasive and have significant mortality.8 Data from a European registry demonstrate age of diagnosis is older for more advanced lesions (1.2 years of age for type I versus 3.2 and 3.1 years for types II and III) with a notably poorer 5-year overall survival (92.9% in type I versus 52.4% and 60.7% for types II and III).8 Treatment may consist solely of resection with close follow up in uncomplicated type I lesions (though some centres advocate chemotherapy post-resection) while all patients with type II and type III lesions undergo chemotherapy with surgery timed to optimise likelihood of maximal resection.8 Chemotherapy protocols have been based on those used to treat rhabdomyosarcomas. In select cases, radiotherapy has been used to target residual disease in those with type II or III.8
Congenital lung malformations (CLMs), including CPAMs, bronchopulmonary sequestrations, bronchogenic cysts and congenital lobar emphysema, are frequently identified on prenatal ultrasound with some studies suggesting the incidence may be as high as one in 3,000 pregnancies.9 Comprehensive reviews of the specific characteristics of these lesions have been published elsewhere.10,11 Surgical removal of these lesions is controversial as they may be asymptomatic and certain types, such as congenital lobar overinflation, may regress with time while the long-term nature of asymptomatic CPAMs remain largely unknown. Current trends seem to favour conservative management of asymptomatic congenital cystic lung lesions in infants.12,13 Ng et al. retrospectively reviewed 74 antenatally diagnosed CLMs13 and noted that over a 10-year period, 65 of the 72 live births with CLMs were able to be managed conservatively, with none progressing to malignancy. Based in part on their findings, they proposed an algorithm based on imaging findings, family history and symptoms for management regarding when to proceed to surgical intervention and observation protocols. This algorithm should be used with some caution, however, as it does not include DICER1 testing as a parameter and prior reports have identified a four percent rate of PPB in otherwise characteristically benign lung lesions.14
Another recent study by Feinberg et al. retrospectively looked at 112 cases of PPB and 103 cases of CPAM.15 Using univariate and multivariate logistical regression, they identified a potential algorithm to distinguish type I PPBs from CPAMs based on radiological and clinical evidence. Prenatal detection, hyperinflated regions and/or systemic feeding vessels were more associated with CPAMs as opposed to PPB. PPBs were found to be more associated with mediastinal shift, complex cysts, bilateral or multilobal disease and DICER1 mutations.15 Again, caution should be exercised as PPBs have been identified prenatally.16 Furthermore, the PPB in our patient did have an unusual systemic blood supply (more typical for CPAMs) though did meet some of the other distinguishing criteria for PPB.
A recent review by Baird et al. graded the current evidence with regards to surgical management.17 They recommended with weak agreement (based on moderate quality of evidence) that asymptomatic cystic congenital lung malformations be removed before symptom onset based on the low morbidity of the procedure, the likelihood of infection during the first year of life (10–30% in some studies), potential for compensatory lung growth and possibility of malignancy.17 They also noted that the natural history of CPAMs is still largely unknown and an area in need of further study. Our case reiterates the need for caution with cystic lung lesions, as the diagnosis of PPB is still a pathological diagnosis2,8 and the tumour can arise in both CPAMs and possible extralobar sequestrations, as in our patient. Given the aggressive nature of PPBs, it can quickly progress from a lesion frequently amenable to complete resection with an excellent prognosis to a lesion with a very poor prognosis despite intensive therapy. Our patient has had close follow up with paediatric oncology and thus far has not demonstrated any signs of relapse, which can occur in about 15% of type I PPB cases.