Cystic fibrosis (CF), an inherited disease affecting all races and ethnicities, results from defects in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which leads to dysregulations in the ion and fluid balance across epithelial membranes throughout the body.1–3 The CFTR protein is an anion channel that functions as a transmembrane traffic ATPase which is regulated by cAMP and protein kinase A phosphorylation.4,5 Due to the prevalent nature of the CFTR channel in epithelial cells, CF-causing mutations lead to multisystemic disease processes. At present, an estimated 80,000 people live with CF worldwide, with over 30,000 within the United States.6,7 Because CF is inherited in an autosomal recessive way, significant portions of the population (~10 million in the USA) are asymptomatic carriers of disease-causing mutations.6,8 The most common disease-causing mutation is Phe508del (F508del), a deletion of the phenylalanine at the 508 position of the protein.6 However, over 2,000 mutations or variants have been identified in the CFTR gene, some known to be disease causing, while the impacts of most are uncertain, and some occurring only in a handful of people with CF.9
In order to conceptualize the vast numbers of CFTR genetic variants and the clinical ramifications, disease-causing mutations have been classified into six categories.10–12 Class I mutations cause severely decreased levels of total protein production due to premature termination of transcription, such as in the case of nonsense mutations and certain splice site mutations or deletions. Class II mutations account for a large majority of CF-causing variants and include the common F508del variant. Class II variants are considered processing mutations that cause misfolding of the CFTR protein; cellular clearance of the abnormal protein in the endoplastic reticulum results in a deficient number of mature CFTR protein successfully trafficked to the cellular surface. Class III mutations are known as gating mutations in which the primary defect is the inability of the CFTR channel pore to open appropriately and thus results in lack of ion transfer in spite of appropriate channel location at the cell surface. Class IV mutations are similar to class III mutations and are conduction mutations which have reduced overall conductance across the channel. Class V mutations have decreased total protein production due to mutations with altered promoter regions or splice sites. Class VI mutations have instability of the formed CFTR protein at the membrane and as a result cause increased turnover of functional protein and thus decreased total ion transport.
Since the description of this classification system, it has been recognized that many mutations exhibit more than one type or class of defect.12 Thus a drug that is able to correct any portion of the defects commonly observed in the CFTR protein may be beneficial for more than a single mutation class. More physiologic and clinically meaningful classifications have been proposed and are often used to describe classes of CFTR mutations. For example, residual function mutations are those CFTR variants which enable some CFTR protein to be expressed at the cell surface versus minimal function (MF) mutations where there is little to no CFTR modulator-responsive protein at the cell surface. As a result, a drug that improves function of CFTR channel would be effective in the case of residual function mutations, but would not be effective for the majority of MF mutations.
The hope for a drug therapy that targets CF pathology at the cellular level is a drastic improvement over traditional CF care. Prior to the advent of CFTR modulators, therapeutic interventions were aimed at decreasing and managing the manifestations of the disease. Inhalation of hypertonic saline and dornase alfa thinned sputum to improve airway clearance; antibiotics were used to treat pulmonary infections; pancreatic enzymes were prescribed to replace missing digestive enzymes; and insulin was increasingly required as people with CF eventually developed pancreatic endocrine insufficiency after years of pancreatic duct mucoid plugging, causing islet cell destruction.13 Instead of palliating measures after the development of disease manifestations, modulators target the root cause of CF by correcting the CFTR protein dysfunction at the cellular level.
Single- and dual-combination CFTR modulators
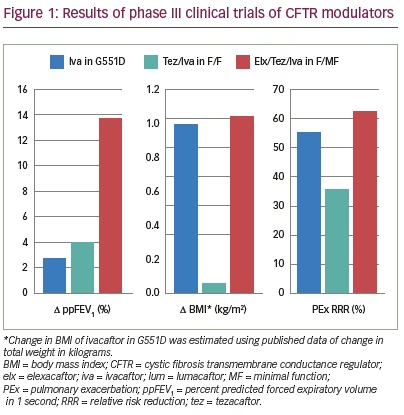
The first clinically available CFTR modulator was ivacaftor (VX-770), initially approved by the US Food and Drug Administration (FDA) in 2012.14 Ivacaftor is a small-molecule potentiator that binds directly to the CFTR protein and increases the time in which the channel is in the open configuration.15–17 While the clinical benefit is most pronounced in people with CF with primary gating mutations, the drug improves chloride transport in a wide variety of CFTR mutations including in the normal protein state.15,18,19 In pre-clinical studies, ivacaftor increased CFTR-dependent ion transport in cultured human bronchial epithelial cells with either G551D or F508del mutations.19 Subsequent clinical studies yielded remarkable results.20,21 In a randomized phase III trial in people with CF with a G551D gating mutation, compared to placebo, ivacaftor dramatically improved the percent predicted forced expiratory volume in 1 second (ppFEV1; +10.6%, p<0.001), respiratory symptom scores as measured by the Cystic Fibrosis Questionnaire-revised (CFQ-R, +8.6 points, p<0.001, minimal clinically important difference = 4.0),22 weight (+2.7 kilograms, p<0.001), and decreased rates of exacerbations (-55%, p<0.001)21 (Table 1 and Figure 1). Since initial FDA approval, ivacaftor’s indication has been expanded to other gating or conductance mutations with residual function of CFTR based on clinical and non-clinical studies.15,23–26 This transformative approach to rapid drug approval speaks to the importance of the strong foundation of understanding of CF pathophysiology, the pre-clinical data in support of clinical benefit, and the sense of urgency to bring highly effective treatments to people with a life-shortening disease.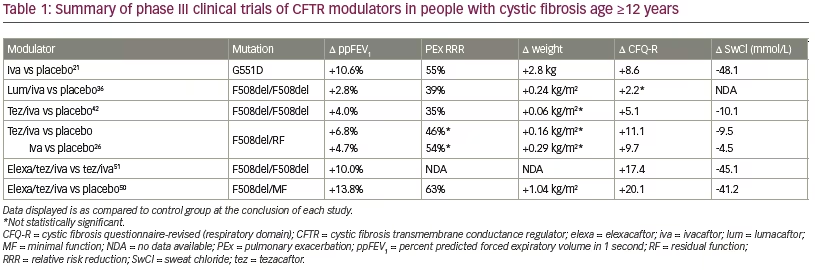
The benefits seen in the initial clinical studies of ivacaftor are sustained over time. People with CF who have been prescribed ivacaftor have significantly lower mortality, need for lung transplantation, hospitalizations, and pulmonary exacerbations over time.27 While ivacaftor proved clearly to be a highly effective therapy, people with CF who could benefit with gating and residual function mutations represented a small fraction (13.6%) of those with CF.6 Unfortunately, the use of a potentiator alone did not improve CFTR function sufficiently in people homozygous for the F508del mutation to provide clinical benefit.28 As a class II mutation, CFTR-F508del results in several protein processing defects, including protein folding, trafficking, and cell surface turnover.29 While CFTR-F508del seems to also have conformational defects leading to gating abnormalities, the singular effect of gating correction is not sufficient to restore function in a clinically meaningful quantity due to a lack of total ion channels successfully transported to the cellular membrane. Although the frequency of the F508del mutation varies across populations, because it is the most common mutation with 84.7% of people with CF in the USA carrying at least one copy, the focus of CFTR modulator development turned to correctors, drugs that could increase the number of ion channels on the epithelial
cell surface.6,30
Lumacaftor (VX-809) was the first CFTR corrector to be approved for clinical use in the USA in combination with ivacaftor in 2015.31 Lumacaftor works by improving CFTR-F508del processing in the endoplasmic reticulum leading ultimately to higher fractions of the defective protein arriving at the epithelial cell surface.32,33 Because CFTR-F508del proteins also exhibit gating defects, lumacaftor in isolation does not rescue the protein in a clinically meaningful way.34,35 However, when used in combination with the gating potentiator ivacaftor, dual drug therapy improved ppFEV1 (+2.6% to +4.0%, p<0.001) and decreased pulmonary exacerbations (-30% to -39%, p<0.001) in people with CF homozygous for F508del.36,37 While these results were not as dramatic as in the case of ivacaftor in those with CF with gating mutations, a 2-year extension study of the trial showed a 42% slower rate of ppFEV1 decline over time.38 As predicted by the relatively modest level of CFTR correction in vitro, the dual combination CFTR modulator did not show efficacy in people with CF heterozygous for F508del and a MF mutation.39
Tezacaftor (VX-661) is chemically similar to lumacaftor and became the next CFTR modulator available, in combination with ivacaftor, to people with CF in the USA (2018).40 As a CFTR corrector, tezacaftor also improves protein folding and trafficking to the epithelial cell surface, but provides the benefit over lumacaftor in not being an inducer of CYP3A4 enzymes and thus exhibits fewer drug–drug interations.41 Additionally, tezacaftor was not associated with increased chest tightness, a problem seen with lumacaftor. Clinical studies of dual combination tezacaftor-ivacaftor show favorable results, with a +4.0% improvement in ppFEV1 (p<0.001) and -35% lower rate of pulmonary exacerbations (p=0.005).42 In people who were heterozygous for F508del and a residual function mutation, including those who were then not eligible for ivacaftor, a parallel group study evaluated the efficacy of tezacaftor-ivacaftor combination and ivacaftor alone in additional mutations shown to be responsive in vitro.39 Tezacaftor-ivacaftor improved ppFEV1 (+6.8%, p<0.001).
While the advent of combination corrector–potentiator modulator therapy served as a proof of concept for the potential to rescue the CFTR-F508del protein, tezacaftor and lumacaftor were not considered highly effective therapies, with only modest clinical benefits when compared to ivacaftor alone in people with responsive gating mutations. Additionally, a significant portion (~42%) of people with CF remained ineligible for any modulator therapy.6,28,39 For a highly efficacious treatment that would impact more people with CF, there was more work to be done.
A triple combination CFTR modulator
In spite of the use of tezacaftor, a more or synergistically effective modulator for trafficking CFTR-F508del protein to the cell surface was needed. The primary target for therapeutic drug development was an effective corrector that would be complementary to the available dual combination therapies. With high throughput drug discovery, several agents were identified as potential targets for development.43
Four next-generation CFTR correctors (VX-152, VX-440, VX-445 or elexacaftor, and VX-659) were evaluated in parallel phase I and II studies in people with CF with either two copies of F508del or one copy of F508del and a MF mutation.43,44 While initial clinical results were remarkably promising for all four compounds, based on efficacy and side effect profiles, VX-445 and VX-659 were selected to be brought to the next stage of clinical testing.43,45–47 The detailed trial design and results of the VX-445 (elexacaftor) and VX-659 preclinical and early clinical studies were published.46,47 In vitro CFTR protein processing and trafficking in airway epithelial cells isolated from people with CF with either F508del homozygous or F508del/MF mutations as assessed by quantity and function of mature CFTR protein, were significantly higher in the presence of VX-445 and VX-659 in combination with tezacaftor and ivacaftor. Both drugs were assessed in two groups of subjects. Critical for consideration of study design, people with CF who were homozygous for F508del mutations had both tezacaftor-ivacaftor and lumacaftor-ivacaftor clinically available for use. Because of the favorable pharmacology (drug–drug interactions), tezacaftor-ivacaftor was chosen as the backbone of triple combination (TC) therapy. In people with CF with F508del and a MF mutation, who had no modulatory therapy clinically available, the investigational drugs in TC with tezacaftor-ivacaftor were compared to placebo. However, in subjects homozygous for F508del mutations, the drug in TC with tezacaftor-ivacaftor was compared to dual combination tezacaftor-ivacaftor as the control.
Clinical studies demonstrated favorable safety and efficacy profiles consistent with the in vitro CFTR activity improvement. In phase II trials, triple therapy with VX-445-tezacaftor-ivacaftor was associated with an increase in ppFEV1 of 13.8% (p<0.001) in the F508del/MF subjects.46 In subjects homozygous for F508del mutations, addition of the third drug VX-445 to tezacaftor-ivacaftor led to an increase in ppFEV1 of 11.0% (p<0.001) on top of that attributable to tezacaftor-ivacaftor alone. Similarly, VX-659-tezacaftor-ivacaftor triple therapy increased ppFEV1 by 13.3% (p<0.001) in the F508del/MF subjects compared to placebo, and by 9.7% (p<0.001) in the F508del homozygous subjects compared to dual tezacaftor-ivacaftor therapy.47 This degree of clinical response to the TC drugs surpassed those of ivacaftor in early clinical trials in people with CF with the G551D mutation, sparking much enthusiasm in the CF community.
In rapidly enrolled phase III studies, VX-659 and VX-445 were evaluated in TC against placebo in subjects with the F508del/MF mutations and against tezacaftor-ivacaftor in subjects homozygous for F508del: AURORA (NCT03525444, NCT03525548 and NCT03525574) and ECLIPSE (NCT03447249, NCT03460990 and NCT03447262).43 In both protocols, the TC was evaluated over 24 weeks in subjects with F508del/MF mutations with the recruitment goal powered to evaluate for significant differences in both changes in ppFEV1 at 4 and 24 weeks, as well as pulmonary exacerbation rates through 24 weeks. However, because USA subjects homozygous for F508del had available dual combination CFTR modulators, the study for this group of subjects was designed for a duration of 4 weeks, powered for change in ppFEV1 only. Because blinded safety data could be obtained from the 48-week studies in subjects heterozygous for F508del/MF mutations, and the change of ppFEV1 at 4 weeks in previous modulator studies has been sustained at 24- and 48-week extension studies, the 4-week endpoint was deemed reasonable by clinicians and the FDA.21,36,42 Placebo and active-controlled arms for both VX-445 and VX-659 were rolled into 96-week open-label extension studies to allow study participants access to drug as well as to gather additional safety data (NCT03525574 and NCT03447262).
The phase III study for VX-659-tezacaftor-ivacaftor was completed first, with interim results released in November 2018 showing a dramatic ppFEV1 improvement of 14.0% (p<0.0001) compared to placebo in subjects with F508del/MF mutations, and a 10.0% (p<0.0001) improvement when VX-659 was added to tezacaftor-ivacaftor in subjects homozygous for F508del mutations at 4 weeks.48 Full study results including incremental improvements in pulmonary exacerbation rates, pulmonary symptom scores, and nutritional status were not available at that time.
VX-445 (elexacaftor)-tezacaftor-ivacaftor performed similarly based on interim results announced in March 2019.49 Subsequently, complete study results were published in both subsets of subjects—those homozygous for F508del and those heterozygous for F508del and a MF mutation. In subjects heterozygous for F508del and a MF mutation without prior modulator therapy available, elexacaftor-tezacaftor-ivacaftor improved ppFEV1 13.8% (p<0.001) at week 4 and 14.3% (p<0.001) at week 24 compared to placebo in 403 subjects.50 The rate of annualized pulmonary exacerbations at 24 weeks was 63% lower compared to placebo (p<0.001). Sweat chloride concentrations improved -41.8 mmol/L compared to placebo at 24 weeks (p<0.001). CFQ-R RD scores at 24 weeks improved 20.2 points compared to placebo (p<0.001). BMI improved 1.04 kg/m2 at 24 weeks (p<0.001), with an absolute increase of 2.9 kg (95% confidence interval 2.3–3.4), as compared to placebo. These results demonstrated that elexacaftor-tezacaftor-ivacaftor was highly effective in subjects with only one F508del mutation.
The TC drug was also evaluated in subjects homozygous for F508del over 4 weeks as compared to dual combination tezacaftor-ivacaftor therapy.51 On top of the impact of tezacaftor-ivacaftor, addition of VX-445 (elexacaftor) improved mean ppFEV1 by 10.0% (p<0.0001), sweat chloride concentration by -45.1 mmol/L (p<0.0001), and CFQ-R RD scores by 17.4 points (p<0.0001). Combined safety events from both patient groups demonstrated that most side effects reported during the study were mild or moderate.50,51 Almost all adverse events were equally reported in the control or placebo groups compared to those receiving the TC drug. The events with a higher signal in the elexacaftor-tezacaftor-ivacaftor arm included diarrhea (13% in TC group versus 7% in placebo), abdominal pain (14% versus 9%), rash (10% versus 5%), and increases in liver enzymes (alanine aminotransferase (ALT), aspartate transaminase (AST), bilirubin: 5–10% versus 1–3%).52 Based on the results of these phase III studies, the FDA approved TC elexacaftor-tezacaftor-ivacaftor through fast-track and orphan drug study status for use in people with CF ≥12 years of age with at least one copy of F508del on October 21, 2019 in a record 62 days after application.52–54
Future directions
For the first time since CF was described by Dr Dorothy Andersen in 1938,55 a highly effective therapy (either ivacaftor alone for those heterozygous for ivacaftor-responsive mutations or elexacaftor-tezacaftor-ivacaftor for those with at least one F508del mutation), which targets the underlying cause of the progressive multi-systemic disease, has the potential to change the clinical course for approximately 90% of people with CF (Figure 2). Unfortunately, a disproportionate number of racial and ethnic minorities do not have an F508 allele and will thus be ineligible for this new therapy.56 Nonetheless, elexacaftor-tezacaftor-ivacaftor is currently being evaluated for use in children with CF aged 6–11 (NCT03691779), and it is expected that demonstration of efficacy and safety in this age group will be followed by testing in younger age groups. It is hypothesized that initiation of highly effective CFTR modulator therapy from the time of diagnosis of CF by newborn screening may prevent the complications of the disease and dramatically change the face of CF.
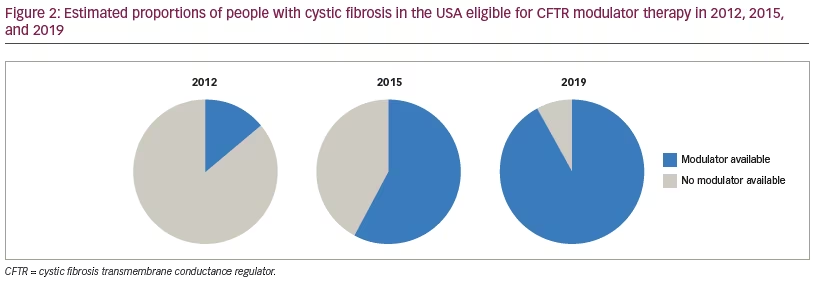
The ~10% of the USA CF population not expected to respond to CFTR modulator therapy predominantly consists of those people with CF with mutations leading to insufficient quantities of mature CFTR protein resulting from premature stop mutations, splice site mutations, and mutations that destabilize the protein at the membrane surface.6,12 Approaches to therapy for this group of people with CF include CFTR amplifiers, read-through therapies, RNA modifying agents, and approaches to direct DNA repair. Clinicians, investigators, and the entire CF community continue to hope that this substantial step forward in the treatment of this life shortening disease will be followed by a cure for all people with CF